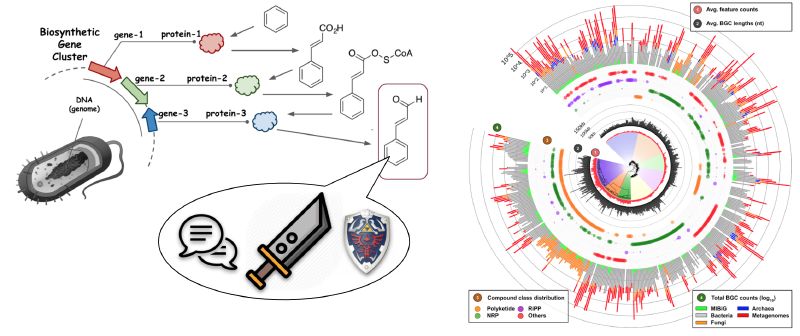
Introduction
随着基因组挖掘技术的进步,生物合成基因簇(BGCs)的研究已成为天然产物发现的核心环节。目前,超过20万个公开的微生物基因组中蕴藏着丰富的化学多样性信息。然而,现有的工具在处理大规模BGCs时,常因基于网络的聚类方法效率低下而面临瓶颈。
BiG-SLiCE(Biosynthetic Gene clusters - Super Linear Clustering Engine) 应运而生,这是一款专为高效聚类大规模BGCs而设计的工具。它通过将BGCs映射到欧几里得空间,以近乎线性的方式将BGCs分组为基因簇家族(GCFs)。在短短10天内,BiG-SLiCE成功分析了来自209,206个微生物基因组和宏基因组组装基因组(MAGs)的1,225,071个BGCs,并构建了全球次级代谢多样性的分布图谱,揭示了未被开发的生物合成潜力。此外,BiG-SLiCE还提供了“查询模式”,能够快速将新测序的BGCs归类到已有的GCFs中,并配备了强大的可视化引擎,便于用户探索数据。这一工具不仅加速了天然产物的发现,还为构建全球可搜索的BGC网络奠定了基础。
BiG-SLiCE的开源地址:https://github.com/medema-group/bigslice

BGCs(以及GCFs)与催化酶及其编码途径产生的化合物直接相关,因此可以作为探索微生物次级代谢化学空间的代理。通过编目所有已测序微生物基因组中的GCFs,可以全面了解现有的化学多样性,并为未来的先导化合物发现提供方向。例如,可以重点关注具有新颖潜力的物种,或识别已知抗生素生产BGC的天然变体。然而,进行此类全球分析需要能够处理海量数据的聚类算法。尽管过去五年中工具的处理能力有所提升(从2014年的11,000–33,000个BGCs到2019年的73,260个),但与当前可用数据量相比仍显不足。截至2020年3月27日,antiSMASH-DB和IMG-ABC两大BGC数据库共包含565,096个BGCs,预测自85,221个细菌基因组。若考虑未覆盖的基因组和宏基因组,这一数字将更大。例如,NCBI RefSeq数据库中的约180,000个细菌基因组可能产生超过1,000,000个BGCs。
处理如此大规模的数据集,即使是目前最快的工具(如BiG-SCAPE)在36核CPU上也需要约37,000小时运行时间,这在实际中几乎不可行。主要瓶颈在于构建相似性网络和聚类分析时使用的成对BGC比较方法,其时间复杂度为O(n²)。因此,迫切需要一种能够更好适应基因组数据增长的替代方法。为此,研究者开发了BiG-SLiCE(Biosynthetic Genes Super-Linear Clustering Engine),它将BGCs映射到欧几里得空间,采用分区聚类算法,时间复杂度接近线性[∼O(n)],从而能够快速分析大规模BGC数据集,实现真正全球化的GCF分析。

为了实现大规模分析,BiG-SLiCE在设计时将可扩展性和速度作为首要目标。与之前的工具BiG-SCAPE(能够敏感捕捉BGCs之间域结构和序列相似性的细微差异)不同,BiG-SLiCE能够在36核CPU、128GB内存的机器上,以不到一周的时间处理超过120万个BGCs的输入数据,同时保持足够的灵敏度以区分输入BGCs中的关键生物合成“信号”。此外,为了便于用户探索和分析结果,BiG-SLiCE还提供了交互式、易于使用的可视化输出,且对软硬件要求极低。
Installation
安装方法非常简单,用以下两种选一个即可
- 从PyPI安装(稳定版)
|
|
- 从源码安装(开发版——仅适用于熟悉操作的用户)
|
|
然后下载最新的HMM模型(约271MB压缩文件)
|
|
如果感觉下载太慢了,也可以手动下载:https://github.com/medema-group/bigslice/releases/download/v2.0.0rc/bigslice-models.2022-11-30.tar.gz,下载后重命名为bigslice_models.tar.gz放到whcih download_bigslice_hmmdb的统一目录下再运行即可,就会跳过下载步骤。
最后检查安装是否成功
|
|
成功安装后,将显示如下信息:
|
|
Usage
BiG-SLiCE 运行模式
[模式1] 聚类分析
解析输入数据集,构建GCF模型(BIRCH聚类),并根据指定阈值(T)进行成员分配。
参数说明:
-i|--input_folder <folder_path>:输入文件夹路径,需包含datasets.tsv文件及数据集子文件夹。--resume:继续上一次的聚类运行(不要与--input_folder同时使用)。--complete:构建GCF模型时,仅使用完整的BGCs(antiSMASH > 4.2 标注为on_contig_edge = False的BGCs)。--threshold:用于GCF模型构建(BIRCH算法)和成员分配的聚类阈值(T)。与--threshold_pct互斥,使用-1关闭此参数(默认:300)。--threshold_pct:基于数据间成对距离的随机采样计算聚类阈值(T),取第N百分位值作为阈值。与--threshold互斥,使用-1关闭此参数(默认:-1)。
[模式2] GCF查询
基于[模式1]生成的GCF模型,从输入文件夹中的BGC GenBank文件中提取特征并进行成员分配。
参数说明:
--query <folder_path>:输入文件夹路径,需包含所有BGC的GenBank文件(支持antiSMASH4的clusterXXX.gbk、antiSMASH5的regionXXX.gbk或MIBiG ≥ 2.0的BGCXXXXXXX.gbk)。--query_name:为查询运行指定唯一名称,便于在输出可视化中追踪。
[模式1+2] 通用参数
适用于聚类和查询模式的参数。
参数说明:
--run_id:指定运行ID进行查询(或继续聚类),而非使用最新运行(可在输出可视化中查看运行ID列表)。--n_ranks <N_RANKS>:为每个BGC的成员分配过程取N个最佳GCF匹配(默认:5)。--program_db_folder <PROGRAM_DB_FOLDER>:HMM库路径(默认:/mnt/local_scratch/kauts001/general-env/bin/bigslice-models)。
CPU/RAM优化
-t|--num_threads:并行运行的作业数(默认:56)。--hmmscan_chunk_size:将biosyn_pfam扫描拆分为每组N个BGCs的块(默认:100)。--subpfam_chunk_size:将sub_pfam扫描拆分为每组N个BGCs的块(默认:100)。--extraction_chunk_size <EXTRACTION_CHUNK_SIZE>:将特征提取拆分为每组N个BGCs的块(默认:100)。--scratch:不将Sqlite3数据库加载到内存中(降低RAM使用,但可能降低速度)。
[Misc] 其他可选参数:
-h, --help显示帮助信息。--export-tsv <folder_path>将现有的预计算输出数据导出为TSV格式文件(指定目标文件夹路径)。--program_db_folder PROGRAM_DB_FOLDER指定HMM库的路径(默认路径:/share/home/jianglab/pengchen/miniconda3/envs/antismash_5.2.0/bin/bigslice-models)。--version显示BiG-SLiCE的版本信息。
Example
可以使用提供的示例输入文件夹进行“最小化”测试运行:
下载https://github.com/medema-group/bigslice/tree/master/misc/input_folder_template这个文件夹,里面只有1个dataset,1个genome,2个gbk。
|
|
这里我遇到了一个问题:
|
|
应该是pyhmmer的版本问题,我尝试安装v0.10.7版本后解决:
|
|
通过以上步骤,可以轻松安装并运行 BiG-SLiCE,开始大规模 BGC 聚类分析!我测试了运行了2min,消耗内存160MB
Input files
为了充分利用BiG-SLiCE的强大功能,其输入的BGC文件需要按数据集和基因组的结构进行组织。一个典型的输入文件夹可能如下所示:
|
|
- datasets.tsv 文件
该文件需要严格命名为 datasets.tsv,并放置在输入文件夹的根目录中。它是一个描述所有BGC元数据的文件,BiG-SLiCE使用它来解析输入文件夹中的所有BGC数据。该文件应该是一个以制表符分隔的文件(.tsv),每一行按以下顺序包含以下内容: • 数据集名称 • 数据集文件夹的路径(相对于输入文件夹根目录的路径) • 分类信息文件路径(见 <taxonomy_X.tsv> 文件) • 数据集描述
以 # 开头的行将被解析器跳过,因此可以用来定义表头等。可以从代码库下载一个模板 datasets.tsv 文件作为起始点。
- <dataset_X> 文件夹
数据集是BiG-SLiCE中用来分组基因组和BGCs的灵活分类方案。例如,可以根据样本来源将Metagenome-Assembled Genomes(MAGs)归为一个数据集,也可以根据原始文献将基因组和MAGs归类,用于元分析研究。每个数据集文件夹下应直接包含基因组文件夹。
- <genome_X> 文件夹与 <genome_X.regionXXX.gbk> 文件
这些文件夹是由antiSMASH运行产生的输出文件夹,包含 <genome_name>.regionXXX.gbk(antiSMASH 5)或 <genome_name>.clusterXXX.gbk(antiSMASH 4)文件。此外,MIBiG >= 2.0格式的文件(名为 BGCXXXXXXX.gbk,可以通过“Download GenBank summary file”获取)也被BiG-SLiCE接受。请确保不修改这些命名格式,因为BiG-SLiCE依赖它们快速区分集群文件(clustergbks)和常规基因组文件。
- <taxonomy_X.tsv> 文件
虽然可以从antiSMASH 5(和MIBiG >= 2.0)生成的cluster genbank文件中提取分类信息(因为这些文件保留了原始基因组的注释),但这些信息并没有统一的方式来为提供的分类名(通常是分号分隔的 ;)指定等级(如:界、门、纲、目、科、属、种等)。为了确保最佳的注释质量和分析结果,BiG-SLiCE要求用户为每个数据集手动提供分类学元数据(如果可能的话),并以制表符分隔的文件(.tsv)格式包含以下内容(按照此顺序):
|
|
为了确保所有数据集中的分类名称一致,建议使用相同的参考数据库为分类名称指定等级。为了帮助用户自动化这一过程,BiG-SLiCE提供了一些Python脚本,可以基于原始输入基因组(而非集群基因组文件)使用GTDB工具包为分类信息赋予等级(此脚本仅适用于完整的古菌和细菌基因组,下载脚本)。如果基因组来自NCBI RefSeq/GenBank(即具有GCF_*或GCA_*号的基因组),可以使用此脚本从GTDB API提取分类信息。
Output
输出文件夹结构:
|
|
直接这样的输出db文件是很难查看的,新版介绍可以用--export-tsv导出表格,
|
|
但是目前有AttributeError: 'tuple' object has no attribute 'values'的报错,官方还没解决。但是可以自己使用sqlite3对结果的data.db文件进行查看和文件导出。
或者使用用户交互输出:
BiG-SLiCE的输出文件夹包含处理后的输入数据(以SQLite3数据库文件的形式)以及一些脚本,这些脚本支持一个小型的Web应用程序来可视化这些数据。要运行该可视化引擎,按照以下步骤操作:
- 安装Web应用的依赖包: 首先,确保安装了所需的依赖包。运行以下命令来安装requirements.txt中列出的所有依赖:
|
|
- 启动Flask服务器: 然后,通过以下命令启动Flask服务器:
|
|
默认情况下,服务器会在端口5000上启动。如果需要自定义端口,可以在命令中指定端口号。
- 打开浏览器访问: 启动服务器后,浏览器将显示如下信息:
Running on http://0.0.0.0:5000/ (Press CTRL+C to quit)
在浏览器中输入 http://0.0.0.0:5000 即可访问Web应用进行数据可视化。
通过上述步骤,您可以轻松启动BiG-SLiCE的可视化应用,查看处理结果和数据分析图表,如下图所示:

BGC Atlas
上次提到过的BGC Atlas,那个数据库用的就是BiG-SLiCE的输出结果。该数据库可供下载的有这几个文件:
-
complete-bgcs.tar.gz A tarball containing the GBK files for only the complete BGC (3.5GB).
-
all-bgcs.tar.gz A tarball containing the GBK files for all BGCs (13GB).
-
bigslice_2.0.0_T0.4_16April.tar.gz A tarball containing the BiG-SliCE clustering of complete BGCs (2.0 GB).
-
atlas_17_07_2004.sql The sql dump of the BGC-Atlas database (2.0 GB).
bigslice_2.0.0_T0.4_16April.tar.gz文件解压后就是BiG-SLiCE的输出文件夹,里面有result文件夹,里面有data.db文件,这个就是BiG-SLiCE处理后的数据库文件。
另外atlas_17_07_2004.sql里面有所有用到的宏基因组metadata,但它是个PostgreSQL database dump,作为文本文件可以直接打开,但不好转换为数据库或是表格,需要用一下postgresql。
使用案例
比如我们自己产生的BGC想要和BGC Atlas进行比较,就可以用[模式2] GCF查询:
|
|
然后就会在big_slice_test_out目录下产生一个report文件夹(如果原来没有的话),report文件夹下可以找到自己本次运行的结果。可以考虑用户交互输出或用数据库软件查看结果。
主要是查看distance(membership_value)这一列,按默认定义的话<0.4才可以认为我们的query和数据库已有的部分聚成了GCF。
然后我们可以查看相应的GCF中有哪些BGCs以及所对应的metadata:
比如我们的BGC和BGC Atlas中的GCF 13734的distance<0.4,可以认为也是GCF 13734的一员,然后快速的方法是去官网查询https://bgc-atlas.cs.uni-tuebingen.de/bgcs?gcf=13734,可以看到一共有258条核心BGC,我们可以下载相应的metadata(一次最多1000条),然后和我们的进行比较分析。
但上述方法只适合查询的BGC不多的情况,很多的话最好还是考虑用代码处理atlas_17_07_2004.sql文件,获取metadata,需要使用postgresql进行处理:
假设你已经会用并成功运行了postgresql(有点难)
|
|
|
|
|
|
然后就可以查看csv并和自己的数据整合分析。
Reference
Satria A Kautsar, Justin J J van der Hooft, Dick de Ridder, Marnix H Medema, BiG-SLiCE: A highly scalable tool maps the diversity of 1.2 million biosynthetic gene clusters, GigaScience, Volume 10, Issue 1, January 2021, giaa154. https://doi.org/10.1093/gigascience/giaa154