Introduction
CRISPR-Cas系统是原核生物中广泛存在的适应性免疫系统,通过识别并切割入侵的噬菌体或质粒DNA/RNA,保护宿主免受外源遗传物质的侵害。自被开发为基因编辑工具以来,CRISPR-Cas系统的研究热度持续攀升,而其天然多样性更是为生物技术创新提供了丰富资源——不同类型的CRISPR-Cas系统(如Class 1的I型、III型,Class 2的II型、V型、VI型)具有独特的组成、作用机制和靶向偏好,例如Cas9(II型)适用于DNA编辑,Cas13(VI型)则针对RNA,Cas12(V型)具有 collateral cleavage活性。
然而,解析微生物基因组中的CRISPR-Cas系统面临两大核心挑战:
- 系统识别的复杂性:CRISPR-Cas系统由Cas基因(构成功能模块)和CRISPR阵列(含重复序列与间隔序列)组成,不同亚型的基因组成差异大,且部分系统可能缺失关键基因(如“孤儿”Cas操纵子或CRISPR阵列),传统注释工具难以完整识别;
- 亚型分类的准确性:早期分类依赖单一特征(如Cas基因或重复序列),易导致误判——例如,相同Cas基因可能对应不同亚型,相似重复序列也可能来自不同系统类型。
发表于The CRISPR Journal的研究“CRISPRCasTyper: Automated Identification, Annotation and Classification of CRISPR-Cas loci”提出了一站式解决方案。该工具整合Cas基因HMM分析、CRISPR阵列检测及机器学习辅助分类,实现了从“基因识别-阵列检测-亚型预测”的全流程自动化,并支持50种CRISPR-Cas亚型(含最新发现的II-C2、II-D、V-M等)的精准分类。本文将简单介绍一下CRISPRCasTyper及其使用方法。
文章介绍
CRISPRCasTyper是一款新型自动化软件工具,用于在原核生物序列中识别和分类CRISPR阵列和cas基因座。该工具基于最新的CRISPR-Cas系统分类和命名法(包含39种亚型/变体),采用机器学习方法对CRISPR阵列的直接重复序列进行亚型分类,能够对孤儿和远缘阵列进行分型。该工具提供图形化输出,以彩色基因图谱形式可视化CRISPR和cas操纵子结构,支持通过同线性分析注释部分和新系统。CRISPRCasTyper在包含31种亚型/变体的手动策划数据集上进行了基准测试,中位准确率达到98.6%。

工具特点与功能
综合识别与分类能力
CRISPRCasTyper能够识别cas操纵子和相关CRISPR阵列,使用HMMER3针对680个隐马尔可夫模型(HMMs)搜索CRISPR-Cas系统相关基因。工具采用两步过滤过程:Class 2效应蛋白(cas9、cas12、cas13)和III-E gRAMP融合蛋白使用特定优化的E值和覆盖率阈值进行过滤,其余Cas蛋白使用E值 cutoff 0.01、序列和HMM覆盖率均为30%的阈值。
机器学习驱动的重复序列分型
作为创新功能,CRISPRCasTyper使用基于梯度提升决策树的机器学习方法,通过对直接重复序列的典型四核苷酸计数进行分析,实现对CRISPR阵列的亚型分类。该分类器在19种包含至少20个已知重复序列的亚型上,对未见测试数据集的中位准确率达到89%。
图形化输出与可视化
工具提供图形化输出功能,以彩色基因图谱形式展示操纵子结构,有助于识别多样化的cas组件或辅助基因,以及部分和新颖的CRISPR-Cas变体/亚型。基因图谱可以扩展包含低于纳入阈值的HMM匹配,这对于发现孤儿CRISPR阵列周围的新系统特别有价值。
处理复杂系统的能力
CRISPRCasTyper能够解析混合CRISPR-Cas系统,并检测跨越环形序列末端的基因座,避免因环形序列的线性表示而导致基因座被错误分割。此外,工具还提供每个检测到的操纵子的干扰和适应模块的完成百分比。
性能基准测试
在与广泛使用的CRISPRCasFinder的比较中,CRISPRCasTyper在识别17种亚型方面表现更优,在其余2种亚型的预测中准确率相当。在这19种亚型中,CRISPRCasTyper的中位准确率为99.5%,而CRISPRCasFinder为93.9%。在所有31种亚型(其中12种在CRISPRCasFinder中缺失)上,CRISPRCasTyper的中位准确率为98.6%。
实施与可用性
CRISPRCasTyper通过conda和PyPi在MIT许可证下提供(https://github.com/Russel88/CRISPRCasTyper),并设有网络服务器版本(http://cctyper.crispr.dk)。网络服务器版本包含一个额外模型,每月使用网络服务器用户输入的众包分型重复序列自动重新训练,确保工具的准确率随时间和使用量的增加而提高。
CRISPRCasTyper提供了最新且免费的软件流程,显著改善了跨基因组序列的CRISPR-Cas基因座自动预测能力。其高效的运行性能(典型基因组2-6 Mbp在4线程下运行时间少于1分钟;深度宏基因组组装60-100 Mbp在20线程下运行时间少于10分钟)使其适用于大规模基因组分析项目。该工具特别适用于处理碎片化的宏基因组组装数据,为研究原核生物防御系统的进化多样性和生态功能提供了强大支持。
CRISPRCasTyper使用教程
官方教程:https://github.com/Russel88/CRISPRCasTyper
CRISPRCasTyper是基于Python 3.8开发的开源工具,支持conda和pip两种安装方式,提供命令行接口(CLI),可实现从数据输入到结果可视化的全流程分析。以下为详细使用步骤(基于GitHub官方文档及研究补充材料)。
CRISPRCasTyper包括了以下50个亚型/变体:
I-A, I-B, I-C, I-D, I-E, I-F, I-F (transposon), I-G, II-A, II-B, II-C, II-C2, II-D, III-A, III-B, III-C, III-D, III-E, III-F, IV-A1, IV-A2, IV-A3, IV-B, IV-C, IV-D, IV-E, V-A, V-B1, V-B2, V-C, V-D, V-E, V-F1, V-F2, V-F3, V-F (the rest), V-G, V-H, V-I, V-J, V-K, V-L, V-M, VI-A, VI-B1, VI-B2, VI-C, VI-D, VI-X, VI-Y.


1. 准备与安装
(1)环境要求
- Python版本 ≥ 3.8
- 依赖工具:HMMER ≥ 3.2(用于Cas基因HMM比对)、Prodigal ≥ 2.6(用于ORF预测)、minced(用于CRISPR阵列检测)、BLAST+(用于重复序列比对)
- 依赖Python包:numpy、pandas、scikit-learn、xgboost、matplotlib、biopython(完整依赖见setup.py)
(2)安装方式
推荐使用conda安装(自动解决依赖冲突并下载数据库),pip安装需手动配置数据库。
方式1:conda安装(推荐)
|
|
方式2:pip安装(需手动配置数据库)
|
|
2. 输入文件与数据要求
CRISPRCasTyper仅需1个输入文件:核苷酸序列的FASTA文件(.fasta或.fa格式),支持以下数据类型:
- 完整基因组(如细菌染色体、质粒);
- 基因组草图(如scaffold或contig);
- 宏基因组组装序列(需用–prodigal meta参数);
- 噬菌体或病毒基因组。
数据预处理注意事项
- 序列格式:FASTA文件中每个序列需有唯一ID(如>CP000816.1,避免特殊字符);
- 序列长度:无严格限制,但短于500bp的序列可能无法检测到完整系统(建议过滤<200bp的contig);
- ** circular基因组**:若输入为环状序列(如完整质粒),需添加–circular参数,避免末端基因被误判为不完整。
3. 核心功能使用
CRISPRCasTyper的核心命令为cctyper,基本语法为:
|
|
以下为不同场景的常用命令示例:
(1)基础分析:完整基因组的CRISPR-Cas解析
以大肠杆菌完整基因组(ecoli_genome.fasta)为例,输出结果到ecoli_output目录:
|
|
(2)宏基因组或短contig分析
宏基因组组装序列通常含大量短contig,需调整Prodigal模式(–prodigal meta)以适配:
|
|
(3)RepeatTyper:仅预测CRISPR重复序列亚型
若仅需分析CRISPR重复序列(无需Cas基因),可使用内置的RepeatTyper工具:
|
|
4. 结果解读与可视化
CRISPRCasTyper的输出目录含多个文件(仅当有对应数据时生成),核心文件及解读如下:
(1)核心结果文件
| 文件名称 | 作用 | 关键列解读 |
|---|---|---|
| CRISPR_Cas.tab | 完整CRISPR-Cas位点信息 | Contig(序列ID)、Operon(操纵子ID)、Prediction(共识亚型)、CRISPRs(关联阵列) |
| cas_operons.tab | 所有Cas操纵子(含孤儿操纵子) | Start/End(操纵子位置)、Prediction(亚型)、Complete_Interference(干扰模块完整性,%) |
| crisprs_all.tab | 所有CRISPR阵列(含可疑阵列) | Consensus_repeat(共识重复序列)、N_repeats(重复次数)、Prediction(关联亚型) |
| crisprs_orphan.tab | 孤儿CRISPR阵列(无相邻Cas操纵子) | 同crisprs_all.tab,新增“Trusted”列(是否为高可信度阵列) |
| spacers/*.fa | 间隔序列FASTA文件 | 每个CRISPR阵列的间隔序列,可用于后续靶标分析(如预测噬菌体靶标) |
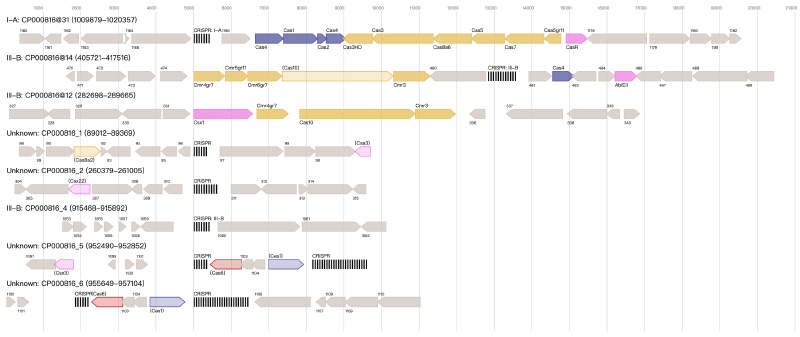
(2)结果可视化:自动生成基因图谱

CRISPRCasTyper会自动生成CRISPR-Cas系统的矢量图(.svg格式),便于直接用于论文,图中颜色含义:
- 黄色:干扰模块基因(如Cas3、Cas9);
- 蓝色:适应模块基因(Cas1、Cas2);
- 红色:Cas6(负责重复序列加工);
- 紫色:辅助基因;
- 灰色:未知基因(编号对应genes.tab);
- 黑白交替块:CRISPR阵列(块数=重复次数)。
示例命令:生成扩展5kb范围的基因图谱
|
|
(3)常见问题与解决方案
- 无结果文件生成:检查输入FASTA格式(是否有唯一ID、序列是否为核苷酸),或降低E-value阈值(–eval 1e-3);
- 内存不足:宏基因组数据可拆分输入文件(如用pyfasta拆分contig),或减少线程数(–threads 2);
- 亚型预测概率低(<0.75):视为不确定结果,可手动检查CRISPR重复序列(crisprs_all.tab的Consensus_repeat列)或Cas基因组成(cas_operons.tab的Genes列)。
References
- Russel, J., Pinilla-Redondo, R., Mayo-Muñoz, D., et al. (2020). CRISPRCasTyper: Automated Identification, Annotation and Classification of CRISPR-Cas loci. bioRxiv, https://doi.org/10.1101/2020.05.15.097824.
- CRISPRCasTyper GitHub仓库:https://github.com/Russel88/CRISPRCasTyper
- Makarova, K. S., et al. (2020). Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nature Reviews Microbiology, 18(11), 675-691.
- Russel, J., et al. (2022). Type IV CRISPR–Cas systems are highly diverse and involved in competition between plasmids. Nucleic Acids Research, 50(11), 6255-6267.