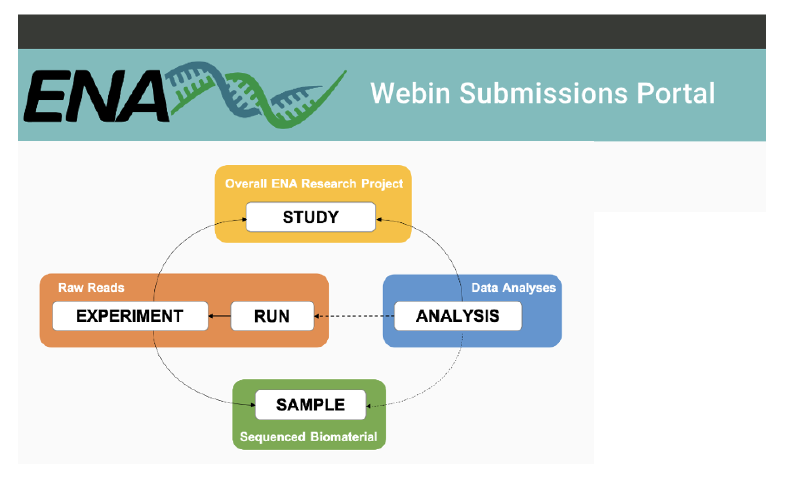
Introduction
很早之前写了一个上传测序数据到ENA的小教程,但是用的人不多,大部分研究还是直接上传到NCBI,会自动同步到ENA。
但最近因为资金短缺,NCBI各种业务暂停维护,但大家投文章又很着急满足杂志社的数据公开政策,ENA就成了重要的一个选择(上传到国内的CNGB有些杂志和审稿人不认可,唉。。。),所以在此更新一下上传的步骤。
European Nucleotide Archive (ENA) 是一个由欧洲分子生物学实验室 (EMBL) 欧洲生物信息学研究所 (EBI) 维护的公共数据库。ENA 收集、存储和提供核酸序列数据,是全球生物学和生物信息学研究的重要资源之一。以下是 ENA 的几个关键点:
-
数据收集和存储:ENA 接收并存储来自全球各地的核酸序列数据。这些数据包括DNA和RNA的序列,以及相关的元数据,如序列的来源、生物学特征和实验信息。
-
数据访问:ENA 提供公开访问接口,允许研究人员和公众免费下载和检索数据。用户可以通过网页界面、编程接口 (API) 和批量下载工具访问数据。
-
数据整合:ENA 与其他主要的生物序列数据库(如美国的GenBank和日本的DDBJ)合作,确保数据在全球范围内的整合和共享。这种合作被称为国际核酸序列数据库协作 (INSDC)。
-
数据提交:研究人员可以向 ENA 提交他们的核酸序列数据。ENA 提供了详细的指南和工具,帮助研究人员提交数据并确保数据的质量和一致性。
-
用途广泛:ENA 中的数据被广泛用于各种生物学研究领域,包括基因组学、转录组学、宏基因组学和进化生物学等。
Submission
由于项目需要,我们要把测序原始数据上传至ENA数据库。在此以宏基因组的双端fastq测序文件的提交流程为例,讲解具体步骤。
官方教程: https://ena-docs.readthedocs.io/en/latest/submit/general-guide.html
在 European Nucleotide Archive (ENA) 提交数据时,现在还是有三种不同的提交方式,每种方式适用于不同类型的数据提交。 根据你的需求,可能需要使用一种或多种提交方式:
-
交互式提交 (Interactive Submissions):
- 方法:通过在浏览器中直接填写网页表单,并下载可以离线填写的模板电子表格,然后将其上传到 ENA。
- 适用场景:这是最易于访问的提交方式,适合大多数用户,尤其是那些不熟悉命令行或编程的用户。
- 优点:操作简单,界面友好,适合初次提交或数据量较少的提交者。
-
命令行提交 (Command Line Submissions):
- 方法:使用 ENA 的专用程序 Webin-CLI 进行提交。这个工具可以在提交之前完全验证你的数据,确保数据的准确性和完整性。
- 适用场景:适合需要高控制力和数据验证的用户,特别是有一定技术背景的用户。
- 优点:提供最大化的控制和数据验证,适合批量提交和大规模数据。
-
编程提交 (Programmatic Submissions):
- 方法:通过准备 XML 文档来完成提交,可以使用 cURL 程序或 Webin Portal 将数据发送到 ENA。
- 适用场景:适合需要自动化提交的用户,尤其是那些有编程能力并且需要频繁提交数据的用户。
- 优点:支持自动化和高效的数据提交流程,适合大规模数据和高频率的提交需求。
我们这里使用的是1+2方法,这样是最通用,灵活且方便的流程。 使用网页注册project和sample,再使用Webin-CLI提交数据。
Install Webin-CLI
使用 Webin-CLI 提交数据到 European Nucleotide Archive (ENA) 需要按以下步骤进行。确保你已经安装了 Java 1.8 或更高版本,并下载了最新版本的 Webin-CLI。
- 步骤 1: 安装 Java
如果你还没有安装 Java,请从以下链接下载并安装 Java 1.8 或更新版本: 下载 Java
检查版本:
|
|
- 步骤 2: 下载 Webin-CLI
从 GitHub 下载最新版本的 Webin-CLI,现在是9.0.1了(2025-11-11): 下载 Webin-CLI
在终端或命令行中使用 java 命令运行 Webin-CLI。
运行 Webin-CLI 并显示帮助信息:
|
|
|
|
| 参数 | 说明 |
|---|---|
-context=TYPE |
提交类型。可选值包括:genome(基因组)、transcriptome(转录组)、sequence(序列)、polysample(多样本)、reads(测序读段)、taxrefset(分类参考集)。 |
-manifest=FILE |
manifest 文件路径(文本文件),其中包含文件路径及元数据字段。 |
-userName, -username=USER |
Webin 提交账户名或邮箱地址。 |
-password=PASSWORD |
Webin 提交账户的密码。 |
-passwordFile=FILE |
包含 Webin 账户密码的文件。 |
-passwordEnv=VAR |
环境变量名,变量中存储 Webin 账户密码。 |
-inputDir, -inputdir=DIRECTORY |
manifest 文件中声明的文件所在的根目录。默认使用当前工作目录。 |
-outputDir, -outputdir=DIRECTORY |
输出文件的根目录。输出目录的结构为:<context>/<name>/<validate,process,submit>。默认使用 manifest 文件所在目录。<name> 为 manifest 文件中定义的唯一名称。验证报告(validation reports)将写入 <validate> 子目录。 |
-centerName, -centername=CENTER |
仅对于代理账户(broker accounts)是必填项。 |
-validate |
仅验证文件,不上传、不提交。 |
-validateFiles |
所有 manifest 字段变为可选项,仅用于验证测序文件,无需提供元数据。 |
-submit |
验证、上传并提交文件。 |
-test |
使用测试提交服务(test submission service)。 |
-ascp |
若系统安装了 Aspera CLI 并在 PATH 中,可使用 Aspera 代替 FTP 上传文件。 |
-help |
显示帮助信息并退出。 |
-fields |
显示所有或特定 context 支持的 manifest 字段。 |
-version |
输出版本信息并退出。 |
-sampleUpdate |
若样本已存在,则更新该样本信息。 |
- 退出代码(Exit codes)
| 代码 | 含义 |
|---|---|
0 |
成功(SUCCESS) |
1 |
内部错误(INTERNAL ERROR) |
2 |
用户错误(USER ERROR) |
3 |
验证错误(VALIDATION ERROR) |
Reads submission
准备好软件后,我们就可以开始提交流程了:

Stage 1: Pre-register Study and Sample
首先进入官网The European Bioinformatics Institute < EMBL-EBI,这里一般实验室有自己的账号,登陆。
我们会进入这个页面:

- 注册study
点击Register Study,按要求填写研究名,计划的释放时间(后续也可以更改),保存好我们的project id文件。里面有形如“PRJEB76945”的Project id,我们一般也是把这个id放在文章的data availability里面即可。
- 注册sample
点击Register Samples,然后“Download spreadsheet to register samples”。

这一步要根据自己数据去下载一个checklist(tsv),用excel打开按要求填好每一个sample的信息,注意不要删掉本来的内容。比如:
- taxid:可在这里查询https://www.ebi.ac.uk/ena/browser/view/408169?show=tax-tree。比如我们是环境样本,空气就是Taxon: 655179
- scientific_name:air metagenome
- sample_alias, sample_title, sample_description, project name
- 有些是有填写格式的,比如时间,经纬度要注意
注意看看每一列的说明,关键是Validation,要确保填的内容满足需求,比如“collection date”这列会用下面的正则表达式验证:
|
|
说明我们要么填严格的时间格式,要么填“not applicable”等等。填完后如下

把这个tsv点“Upload filled spreadsheet to register samples”上传回去,系统会检查有效性,如果有报错就按照要求重新修改下。最后提交好后就有每个sample的ACCESSION了。
Stage 2: Prepare the Files
首先测序原始fastq文件需要进行压缩(.fq.gz):
|
|
manifest files
作为提交的一部分,每个刚刚注册的Sample(一个单端测序文件,或一对双端测序文件)都必须使用一个清单manifest文件描述。
manifest文件使用Webin-CLI的-manifest <filename>参数指定,每个manifest文件需包含以下内容:
- 通用实验信息
- Study accession or unique name (alias)
- Sample accession or unique name (alias)
- Experiment name
- Sequencing platform
- Sequencing instrument
- Library name (optional)
- Library source
- Library selection
- Library strategy
- Free text library description (optional)
- Insert size for paired reads (optional)
- 测序文件,以下的一项
- BAM file
- CRAM file
- Single Fastq file
- Paired Fastq files
- Multi-fastq files
单独填写每一个样本的manifest文件比较麻烦,可以用一段简单的R脚本帮助编写:
首先,根据我们之前注册时已经得到的信息整理出一个excel(列名不要改变,FASTQ这列如果是双端就用; 隔开):

|
|
这样会在manifest文件夹下生成每一个样本的manifest文件,内容如下:
|
|
Stage 3: Validate and Submit Files
终于到了上传的这一步了,ENA建议在上传前先用-validate验证清单文件中定义的文件(只验证不提交)。
我们拿一个样本试一试,填自己的用户名和密码,没问题:
|
|
正式提交,用for循环提交每一个样本(-submit也会在提交前验证):
|
|
上面那个命令validate可以执行并通过,但默认是ftp协议传输,如果你的提交没问题那就跳过下面ascp这部分:
-ascp submit
我使用默认的ftp好像有点问题,进度条都没有,应该是我们服务器的问题。
那就试一下-ascp参数,但是我之前装好的ascp不能用了,好像是最近aspera改过一次:
|
|
没办法,重装,官网下载安装没用,应该是服务器配置不匹配。
只能试试conda,conda install -c hcc aspera-cli -y。
虽然很慢,但最后conda安装的可以用。
加上-ascp参数,大功告成,上传速度也非常快:
|
|
最后,我们可以回到官网页面:The European Bioinformatics Institute < EMBL-EBI。点击左侧的Dashboard,再点击Raw Reads里的Runs Report,就可以查看每一个样本提交状态了,没问题:

在所有文件成功提交后,等到我们设定的释放时间,project就会自动公开,可以搜索到项目页面。但按照我之前上传的经验,大概过几天后(可能后台系统审核什么的),对应的reads才会公开出来。
Assemblies submission
上传每个宏基因组样本的assembly(Primary Metagenome,Binned Metagenome和MAG)方法也类似,前面的注册Study和Sample相当于我们已经完成了,不过最后的Manifest文件的内容跟raw reads data不太一样。
这里讲一下我们经常会组装得到的contigs和binning得到MAGs上传方法。
Contigs
所谓“初级宏基因组组装”(primary metagenome assembly)指的是:尚未进行分箱 (binning),直接从一个样本生境或多个样本生境集合中拼出的宏基因组组装,没有尝试拆分不同物种/分类群。
第 1 阶段:预注册研究和环境样本
每次提交必须关联一个已预注册的研究 (study) 和一个环境样本 (environmental sample)。该样本应当与用于生成你的原始读段 (raw reads) 的样本一致。
在注册环境样本时,请保证选择了适当的环境检查表 (environmental checklist),并使用一个环境分类号 (environmental taxon)(例如水生宏基因组 aquatic metagenome,tax id: 1169740) 关于研究/样本注册的方法与其他研究/样本注册流程相同。请参考其对应链接。
强烈建议你同时提交并引用用于生成组装的原始读段 (raw reads)。为关联这些读段,请在 manifest 文件中使用 RUN_REF 标签。这里填我们上面上传reads时对应样本的run accession即可。
第 2 阶段:准备文件
提交所需的文件通过 manifest 文件指定。使用 -manifest <filename> 选项来提供该文件。
一次初级宏基因组组装提交包括以下文件:
- 一个 manifest 文件
- 一个 FASTA 格式的文件
Manifest 文件
该 manifest 文件为两列格式,以制表符 (tab) 或任何空白字符分隔:
- 第一列:字段名(field name,大小写不敏感)
- 第二列:字段值 (field value)
以下是支持的 metadata 字段:
STUDY:研究 accession 或唯一别名 (alias)SAMPLE:环境样本 accession 或唯一别名 (alias)ASSEMBLYNAME:唯一的组装名称ASSEMBLY_TYPE:填 “primary metagenome”COVERAGE:估计的测序覆盖深度PROGRAM:拼装程序名称PLATFORM:测序平台,或以逗号分隔的多个平台MINGAPLENGTH:最小连续 N 长度被视为缺口 (optional)MOLECULETYPE:选 “genomic DNA”, “genomic RNA” 或 “viral cRNA” (optional)DESCRIPTION:组装的自由文字描述 (optional)RUN_REF:逗号分隔的运行 (run) accession 列表 (optional)
以下是支持的文件名字段:
FASTA:以 FASTA 格式提供序列文件
示例 manifest 内容:
|
|
第 3 阶段:验证并提交文件
提交命令:
注意要通过参数 -context genome,其他命令参数一致。
|
|
-
组装名称验证 (Assembly name validation)
- 匹配正则模式
^[A-Za-z0-9][A-Za-z0-9 _#-.]*$(即首字符为字母或数字,后续字符可包括字母、数字、空格、下划线、井号、减号或点号) - 不超过 50 个字符
- 不能包含被拼装生物体的分类学名称
- 匹配正则模式
-
序列验证 (Sequence validation)
- 序列名称在一个组装内必须是唯一的
- 序列长度至少要 20 bp
- 序列末端不能有 N 字符
- 序列只能由以下碱基字符组成:‘a’, ‘c’, ‘g’, ‘t’, ‘u’, ‘b’, ‘d’, ‘h’, ‘k’, ‘m’, ‘n’, ‘r’, ‘s’, ‘v’, ‘w’, ‘y’
MAGs
在 ENA 中,Metagenome-Assembled Genome(MAG)定义为:基于一个或多个已分箱(binned)的宏基因组(metagenome)所得的、单一分类群(single-taxon)组装,该组装被断言为接近一个真实个体基因组(可能匹配已有的纯培养菌株,或者代表一个新发现的菌株)。
MAG 提交按照与纯培养/分离菌株基因组相同的层级处理,并在 INSDC 中以相同方式分布。 由于环境样本中可能存在多个同种的基因组重复,而且 MAG 组装更容易受到污染(contamination)影响,因此我们要求只提交每个物种在其生态系统(biome)中最高质量、唯一代表的 MAG。
建议在提交 MAG 前,先提交所有的低一级组装(即先提交原始读段、初级宏基因组组装、分箱结果等)。这样 MAG 提交可以突出显示从分箱宏基因组集合中派生出的“最佳代表”组装,并且确保所用方法具有可复现性。
基因组组装提交(genome assembly submissions)可包括:质粒(plasmids)、细胞器(organelles)、完整病毒基因组(complete virus genomes)、病毒片段/复制子(viral segments/replicons)、噬菌体(bacteriophages)、原核(prokaryotic)和真核(eukaryotic)基因组。 一个 MAG 通常包括以下内容:
- 通用组装信息,例如:研究 (study) accession 或唯名 (alias)、MAG 样本 accession/唯名、组装名称 (assembly name)、组装程序 (assembly program)、测序平台 (sequencing platform)、最小缺口长度 (minimum gap length)、分子类型 (molecule type)、测序覆盖度 (coverage)、自由文本描述 (optional)。
- Contig/Chromosome 序列
- (可选)功能注释 (functional annotation) 注:在此文档中,“chromosome”一词可理解为包括细胞器、质粒、病毒片段。
第 1 阶段:预注册研究、样本及低级组装
每次 MAG 提交必须关联一个已预注册的研究 (study) 和一个 MAG 样本 (MAG sample)。 在注册研究 (study) 时,如果你打算提交注释(annotation)内容,需确保在此阶段注册基因座标签前缀 (locus tag prefixes)。 建议你在提交 MAG 前,先提交所有低一级的宏基因组组装和原始读段,以便记录方法并提高可复现性。
每个 MAG 组装提交必须关联一个 MAG 样本。原因在于:MAG 并不是采自环境样本中全部数据的组装,将其挂在环境样本下可能引起错误的分类学归属 (taxonomy assignment)。
这些 MAG 样本代表由环境样本派生出来的单个有机体 (organism) 子集,并承载其所有与分类学、方法相关的元数据。
注册时,MAG 样本应尽可能给出具体分类学信息,并使用 Genomic Standards Consortium (GSC) 的 MIMAGs (checklist) 规范。
请确保 MAG 样本正确地引用其派生自哪一个环境样本,例如:
“This sample represents a MAG derived from the metagenomic sample ERSXXXXX” 如果你仅提交 MAG 而不提交其低级组装或读段,也应注册一个环境样本,并在 MAG 样本中引用该环境样本。
第 2 阶段:准备文件
提交所需的文件通过一个 manifest 文件指定。使用 -manifest <filename> 参数。
提交 MAG 所需文件依组装层级 (assembly level) 而定,常见层级为 “Contig assembly” 或 “Chromosome assembly”。
Contig Assembly层级包括:
- 一个 manifest 文件
- 一个 FASTA 文件 或者 一个 flat file(EMBL-Bank 格式) 若序列数少于 2 或多于 1,000,000 会产生错误。若你的组装仅为单一序列且质量很高,应考虑提交为 Chromosome assembly。若未达到完全组装染色体标准但认为质量足够提交为 MAG,也可向帮助台申请标为 ‘single contig’ 组装。
Chromosome Assembly层级包括:
- 一个 manifest 文件
- 一个 FASTA 文件 或者 一个 flat file
- 一个 chromosomes list 文件
- 可选 0-1 个 unlocalised list 文件(当有未定位序列时)
序列名在一个提交中必须唯一,并且在 FASTA/flat 文件、chromosome list 文件中应一致引用。
Manifest 文件
manifest 文件为两列格式(字段名/值),用制表符或空白分隔。字段名称不区分大小写。 支持的 metadata 字段包括:
STUDY:研究 accession 或唯一别名(alias)SAMPLE:MAG 样本 accession 或唯一别名(alias)ASSEMBLYNAME:唯一的组装名称ASSEMBLY_TYPE:填写 “Metagenome-Assembled Genome (MAG)”COVERAGE:估计的测序覆盖度PROGRAM:组装程序名称PLATFORM:测序平台/多个平台用逗号分隔MINGAPLENGTH:视为缺口的最小连续 N 长度 (optional)MOLECULETYPE:可选,“genomic DNA”, “genomic RNA”, 或 “viral cRNA”DESCRIPTION:组装的自由文本描述 (optional)RUN_REF:逗号分隔的运行 (run) accession 列表 (optional)
支持的文件名字段包括:
FASTA:FASTA 格式序列文件FLATFILE:EMBL-Bank 格式 flat 文件AGP:AGP 格式文件(如果适用)CHROMOSOME_LIST:染色体清单文件(仅 Chromosome assembly)
示例 manifest 文件 :
|
|
第 3 阶段:验证并提交文件
提交命令示例:
注意要通过参数 -context genome,其他命令参数一致。
|
|
分配编号 (Assigned accession numbers)
一旦组装提交完成,通过 Webin-CLI 会立即分配一个分析 (analysis) accession,格式为 ERZxxxxxx,提交者可即时获取。 但 ERZ accession 不应用于论文中引用。它主要用于提交者在 Webin 系统中引用其提交。 可用于长期稳定引用的 accession 包括: * 研究 accession(如 PRJEBxxxxx) — 在研究注册时分配。 * 样本 accession(如 SAMEAxxxxxx) — 在样本注册时分配。 * 基因组组装 accession(如 GCA_xxxxxxx) — 在组装完全处理后分配。 * 序列 accession(s) — 在组装完全处理后分配。
- 样本与研究验证 (Sample and Study Validation)
- 一个研究 (study) 与一个样本 (sample) 的配对必须是唯一的(一个组装对象不能重复使用相同研究+样本已被用过)。
- 样本的分类学归属必须在 NCBI 分类树中为 物种级别或以下(species rank 或下一级)或等效级别。
- 组装名称验证 (Assembly name validation)
- 名称必须满足正则表达式:
^[A-Za-z0-9][A-Za-z0-9 _#-.]*$(即首字符为字母或数字,后续字符可为字母、数字、空格、下划线、井号、减号或点号)。 - 长度不得超过 50 字符。
- 不能包含被组装生物体的分类学名称。
- 名称必须满足正则表达式:
- 染色体名称验证 (Chromosome name validation)
- 名称必须满足正则表达式:
^[A-Za-z0-9][A-Za-z0-9_#-.]*$。 - 长度必须少于 33 字符。
- 名称中(不区分大小写)不得包含以下任何词: “chr”, “chrm”, “chrom”, “chromosome”, “linkage group”, “linkage-group”, “linkage_group”, “plasmid”。
- 名称必须在本组装中唯一。
- 名称必须满足正则表达式:
- 序列验证 (Sequence validation)
- 序列名称在一个组装内必须唯一。
- 序列长度至少为 20 bp。
- 序列末端不能有 ‘N’ 字符。
- 序列只能由以下碱基字符组成: ‘a’, ‘c’, ‘g’, ‘t’, ‘u’, ‘b’, ‘d’, ‘h’, ‘k’, ‘m’, ‘n’, ‘r’, ‘s’, ‘v’, ‘w’, ‘y’。