Introduction
题为“Global biogeography of airborne viruses in public transit systems and their host interactions”的文章近期在《Microbiome》上发表。该研究分析了全球六个城市公共交通系统空气样本的503个宏基因组,旨在明确空气传播病毒的多样性、组成及功能潜力,探究病毒与宿主的相互作用。
- Lei, H., Du, S., Tong, X., Chan, W.L., Leung, M.H.Y., Bøifot, K.O., Bezdan, D., Butler, D.J., Danko, D.C., Green, D.C., et al. (2025). Global biogeography of airborne viruses in public transit systems and their host interactions. Microbiome 13, 193. https://doi.org/10.1186/s40168-025-02173-z.
可以学习参考一下其中从宏基因组数据分析病毒的详细流程。
建筑环境(BEs)空气中虽营养匮乏且受物理、环境条件波动影响,但仍存在大量细菌、真菌和病毒。数据显示,空气传播细菌与真菌平均浓度约为1×10⁵个/立方米,病毒浓度与之相近。不过,目前对细菌和真菌的组成及代谢功能研究较多,对空气传播病毒的认知却十分有限。
过往研究发现,不同室内空间有独特的空气传播病毒群落,人乳头瘤病毒和多瘤病毒较常见;机械通风场所中,病毒还呈现季节性动态变化,说明通风率、室外天气等季节相关因素会影响病毒组成。病毒能依环境调整生活方式,导致自身及宿主丰度波动,尤其毒性噬菌体引发宿主细胞裂解时更为明显。“Kill-the-Winner”(杀死获胜者)和“Piggyback-the-Winner”(搭便车获胜者)等生态模型,可描述病毒与宿主丰度的平衡关系,助力理解病毒捕食策略及相互作用的复杂性。同时,病毒进化出携带辅助代谢基因(AMGs)的能力,噬菌体编码的AMGs能增强或改变宿主代谢以提升自身适应性;而微生物宿主为抵御病毒,进化出CRISPR-Cas系统,病毒则进一步进化出抗CRISPR(Acr)蛋白应对。
在富营养环境中,噬菌体AMGs可调控宿主代谢途径,CRISPR-Cas系统与Acr蛋白也会协同进化。例如,污水处理系统中可见微生物宿主表达噬菌体AMGs,暗示其在污染物去除中起作用;人体肠道内,病毒通过间隔序列与微生物宿主关联,符合“红皇后假说”的协同进化过程。
尽管低营养环境(如海洋、沙漠)中病毒的宿主捕食策略及协同进化已有研究,但建筑环境这类存在持续气流的场景,相关研究仍空白。因此,该研究分析全球六个城市(丹佛、香港、伦敦、纽约、奥斯陆、斯德哥尔摩)公共交通系统空气样本的503个宏基因组,旨在明确空气传播病毒的多样性、组成及功能潜力,探究病毒与宿主的CRISPR-Cas系统和Acr蛋白协同进化,以及病毒生活方式与宿主丰度的关系,并假设病毒生物学特征具有生物地理分布模式,且病毒-宿主相互作用受建筑环境条件影响。
研究方法
样本采集、基因组DNA提取与宏基因组测序
2018-2019年6-7月,研究人员从六个城市公共交通系统采集503份空气样本。其中丹佛13份来自铁路和公交系统,其余城市样本均来自地铁(香港159份、伦敦76份、纽约96份、奥斯陆127份、斯德哥尔摩32份)。采样在工作日进行,地点建筑特征各异。
采样使用配备驻极体微纤维过滤器的SASS 3100干空气采样器,流量300升/分钟,时间30分钟。采样器置于1.5米高三脚架上,呈45°角朝下倾斜,样本采集后储存于-80°C。同时制备两种阴性对照:现场对照(放置新过滤器但不运行采样器)和实验室对照(对新过滤器提取DNA)。
样本经干冰运输至挪威国防研究机构提取DNA:先将过滤器颗粒物提取到NucliSENS裂解缓冲液,离心分离上清液与沉淀物;沉淀物经酶解和机械裂解释放DNA,用DNeasy PowerSoil试剂盒去除抑制剂后与上清液混合;再用NucliSENS磁性提取试剂试剂盒提取DNA(调整磁性硅悬浮液体积至90μL,孵育20分钟)。
同步处理14个阴性对照和3个阳性对照(ZymoBIOMICS微生物群落标准品),用Qubit 3.0荧光计确定DNA浓度,随后构建测序文库,在HudsonAlpha基因组中心用Illumina HiSeq X系统进行150 bp双端宏基因组测序。
病毒生信分析流程

原始测序数据预处理与质量控制
- 原始读数过滤:使用
Trim Galore(v0.6.10)对 Illumina HiSeq X 产出的 150 bp 双端原始测序数据进行质控,参数设为--length 50 --q 20,即保留长度≥50 bp、质量值≥20 的序列,剔除短片段和低质量片段。 - 人类序列去除:通过
KneadData(v0.7.4)工具,以Genome Reference Consortium Human Build 37人类基因组为参考数据库,过滤样本中混杂的人类基因组序列(采用默认参数),避免人类序列对微生物分析的干扰。 - 阴性对照污染剔除:
- 用
MEGAHIT(v1.1.3)先组装“现场对照(未运行采样器的新过滤器)”和“实验室对照(仅提取新过滤器DNA)”的序列,得到阴性对照重叠群; - 将样本序列与阴性对照重叠群比对,去除可映射到对照重叠群的污染序列;
- 用
- 残留污染识别:
- 用
Kraken2(v2.1.3)对剩余序列进行物种分类注释,Bracken(v2.8)校正分类计数; - 借助 R 包
decontam(v1.12)的“流行度模式”,以 0.1 为概率阈值,识别并通过 Python 脚本extract_kraken_reads.py去除潜在交叉污染序列;
- 用
- 最终清洁数据:经上述步骤后,每个样本平均获得 18.8±7.2 百万条双端清洁读数,用于后续组装。
双路径病毒分箱
-
路径1:宏基因组分箱+病毒解析
- 序列映射与覆盖率计算:用
minimap2(v2.24)将清洁读数比对到样本重叠群(前序步骤中用MEGAHIT组装),通过samtools(v1.6)过滤低质量比对结果; - 宏基因组分箱:用
MetaBAT2(v2.12.1)的jgi_summarize_bam_contig_depths模块计算重叠群覆盖率,再用VAMB(v4.1.3)(默认参数)将重叠群聚类为宏基因组分箱; - 病毒分箱提取:通过
PHAMB(v1.0.1)推荐工作流程,从宏基因组分箱中解析出仅含病毒序列的“病毒分箱”(PHAMB 排除非噬菌体分箱的准确率达 93%-99%,远高于其他工具)。
- 序列映射与覆盖率计算:用
-
路径2:病毒序列识别+分箱优化
- 假定病毒重叠群识别:用
DeepVirFinder(v1.0)(得分>0.5 且 p 值<0.05)和VirSorter2(v2.2.4)(得分>0.5)分别识别样本重叠群中的假定病毒序列; - 病毒分箱生成:用
vRhyme(v1.1.0)对上述假定病毒重叠群进行聚类,生成病毒分箱; - 分箱质量优化:由于
DeepVirFinder(准确率 69%-74%)和VirSorter2(准确率 30%-84%)的假阳性率较高,测试 0.6-0.9 共 4 个更高得分阈值,最终保留“不含路径1中 PHAMB 分箱重叠群”的病毒分箱,避免重复。
- 假定病毒重叠群识别:用
病毒操作分类单元(vOTUs)构建与质量筛选
- vOTUs 聚类:按 95% 平均核苷酸一致性(ANI) 和 85% 比对覆盖率 标准,对两种路径获得的所有病毒分箱进行聚类,每个聚类中最长的序列作为该 vOTU 的代表序列;
- 质量分级与筛选:用
CheckV(v1.0.1)(默认参数)将 vOTUs 分为“完整”“高质量(完整性>90%)”“中等质量(完整性 50%-90%)”“低质量”“未分类”5 级,仅保留前 3 级 vOTUs(共 5346 个); - 有效性验证:用
DeepVirFinder、VirSorter2、VIBRANT(v1.2.0)三种工具交叉验证,确认 82.8% 的 vOTUs 为真实病毒序列。
病毒生活方式预测
通过 VIBRANT(v1.2.0) 的 virome 标记功能判断 vOTUs 生活方式:
- 若 vOTU 含“整合酶类注释”或被识别为“原噬菌体”,则归类为 温和型病毒;
- 无上述特征且被 VIBRANT 确认为病毒的,归类为 毒性型病毒;
- 未被 VIBRANT 识别为病毒的,标记为“生活方式未知”。
病毒分类学注释
- 物种级分类:用
Prodigal(v2.6.3)(参数--p meta)预测 vOTUs 的开放阅读框(ORFs),通过Diamond(v2.6.1)(参数:--evalue 1e-5 --max-target-seqs 10000 --query-cover 50 --subject-cover 50)在IMG/VR 数据库(v4.1)中搜索同源蛋白,若某 vOTU 中≥20% 蛋白质匹配同一物种,则分配该物种分类; - 科级分类(病毒聚类 VCs 构建):
- 合并“所有中等质量及以上 vOTUs”与“NCBI RefSeq 参考病毒基因组(2023 年 5 月,n=16398)”;
- 按 ≥20% 平均氨基酸一致性(AAI)且共享≥8 个病毒蛋白质/≥20% 病毒蛋白质 标准聚类,得到病毒聚类(VCs);
- 若某 VC 中≥20% 参考病毒属于同一科,则为该 VC 分配科级分类;无参考病毒的 VC 标记为“新病毒科”。
病毒丰度计算
- 数据标准化:用
seqtk(v1.4)(参数--s 100)将每个样本的清洁读数稀释至 450 万条(匹配读数最少样本的量),排除 5 个稀释后数据不足的样本; - 映射与丰度计算:用
Bowtie2(v2.5.1)(very-sensitive模式)将稀释后读数比对到 vOTUs,CoverM(v0.6.1)(参数--min-read-percent-identity 95)过滤低一致性比对,再通过CoverM的contig模式(参数--min-covered-fraction 0.7)计算每个 vOTU 的 每千碱基百万映射读数(RPKM); - 相对丰度标准化:某 vOTU 的相对丰度 = 该 vOTU 的 RPKM / 样本中所有 vOTUs 的 RPKM 总和;某 VC 的相对丰度 = 其包含所有 vOTUs 的相对丰度之和。
病毒功能注释
- 核心功能注释:用
HMMER软件包的hmmsearch工具(默认参数),在Kofam、TIGRFAM、Pfam、VOGDB、地球病毒组数据库5 个数据库中搜索 vOTUs 的 ORFs,按 E 值≤10⁻⁵且比特值≥60 保留最优匹配,未匹配 ORFs 归为“功能未知”; - 基因簇聚类:用
MMseqs2(v14.7e284)按 30% AAI 和 70% 比对覆盖率 将 ORFs 聚类为基因簇,基于Pfam注释将功能分为 DNA 结合/调控、裂解、复制、结构、转运蛋白、其他 6 类; - 抗生素抗性基因(ARGs)识别:
- 用
RGI(v5.1.0)(参数--low_quality)搜索CARD 数据库(v3.2.7),NCBI 抗菌药物抗性查找工具(v3.11.14)搜索NCBI 抗菌药物抗性数据库(v3.11),BLAST-蛋白质(一致性 80%、覆盖率 70%)搜索SARG 数据库(v3.0); - 保留 3 个数据库中的独特 ARGs,若亚型注释冲突,以 RGI 结果为准;某 ARG 亚型的相对丰度 = 该城市所有样本中该亚型包含 ARGs 的相对丰度之和;
- 用
- 辅助代谢基因(AMGs)预测:用
DRAM(v1.4.5)的DRAM-v功能(推荐工作流程)检测假定 AMGs,辅助得分 1 或 2 的基因归为 AMGs,用Phyre2(v2.0)(正常建模模式)预测 AMGs 的蛋白质结构(置信度 82%-100%)。
病毒-宿主关联预测
- 体外宿主预测:用
VirHostMatcher-Net(含 62493 个原核基因组的默认数据集),基于病毒与宿主基因组的序列相似性预测体外潜在宿主; - 体内宿主验证:
- CRISPR 间隔序列匹配:从样本宏基因组组装基因组(rMAGs,完整性>50%、污染<10%)中提取 CRISPR 间隔序列,用
BLAST-短核苷酸(E 值≤10⁻⁵、字长 18、一致性≥95%、错配≤1)比对到 vOTUs,建立“间隔序列来源 rMAG-病毒”关联; - 基因组区域匹配:用
BLAST-核苷酸(比特值≥50、E 值≤10⁻⁵、一致性≥96%)将 vOTUs 比对到 rMAGs,映射区域≥1000 bp 的病毒与对应 rMAG 关联;
- CRISPR 间隔序列匹配:从样本宏基因组组装基因组(rMAGs,完整性>50%、污染<10%)中提取 CRISPR 间隔序列,用
- 关联筛选:仅保留“某城市 vOTU 与该城市存在的 rMAG”的关联,用
Cytoscape(v3.10.0)可视化城市级病毒-宿主关联网络。
CRISPR-Cas 系统与 Acr 蛋白分析
- CRISPR-Cas 系统识别:用
CRISPRCasTyper(v1.8.0)(默认参数)从 686 个 rMAGs 中识别 CRISPR-Cas 基因、阵列及亚型(共识别出 I、II、III、V 四种类型,I 型最常见); - Acr 蛋白识别与分类:
- 用
AcaFinder和默认AcrDatabase从 vOTUs 中识别 Acr 同源物,若病毒重叠群含“螺旋-转角-螺旋结构域蛋白”,则确认为 Acr 蛋白; - 用
Diamond(v2.6.1)(E 值≤10⁻⁵、比特值≥60)将 Acr 蛋白比对到AcrDatabase,确定其亚型(主要为 II-C、I-B、I-C 亚型); - 用
Mafft(v7.520)进行多序列比对,FastTree(v2.1.11)(JTT 模型)构建 Acr 蛋白与 339 个实验验证 Acr 参考序列的系统发育树。
- 用
统计与可视化分析
- 多样性分析:用 R 包
vegan(v2.6.4)计算 α 多样性(香农指数、观测物种丰富度)和 β 多样性(布雷-柯蒂斯相异度),通过cmdscale函数对 β 多样性进行主坐标分析(PCoA),可视化病毒群落差异; - 差异检验:两组间差异用 曼-惠特尼检验,多组间差异用 克鲁斯卡尔-沃利斯检验;用
vegan的adonis2函数(置换次数=999,方法=“bray”)进行置换多元方差分析(PERMANOVA),评估城市/年份对病毒组成的影响;用betadisper函数进行置换多元离散度分析(PERMDISP); - 相关性分析:通过 R 包
ggpubr(v0.6.0)的stat_cor函数计算 皮尔逊相关系数 和双侧 p 值,分析病毒丰度与宿主丰度、地理距离的相关性; - 可视化工具:除上述
Cytoscape、PCoA外,用 R 包Rtsne(v0.17)可视化 VC 内 vOTUs 分布,用FastTree构建系统发育树,所有统计检验以 p<0.05 为显著标准。
研究结果
空气传播病毒的生物地理学特征

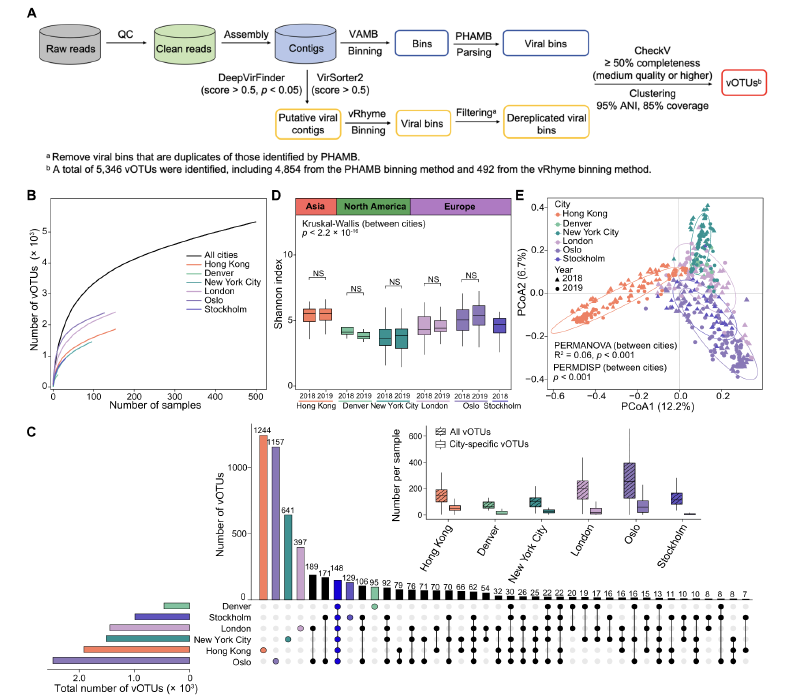
分析503个宏基因组,共获得5346个至少中等质量的vOTUs(含249个原噬菌体),其中3084个为高质量或完整vOTUs,82.8%经验证为病毒。这些vOTUs中,34.3%为毒性型,12.9%为温和型,52.8%生活方式未知。
各城市均有特异性vOTUs(数量7-81个),仅148个vOTUs在所有城市存在。不同城市样本α多样性差异显著(克鲁斯卡尔-沃利斯检验,p<2.2×10⁻¹⁶),香港和奥斯陆样本α多样性高于其他城市,北美城市样本病毒多样性低于其他大洲(曼-惠特尼检验,p<0.01);β多样性分析显示病毒组成按城市和大洲聚类(PERMANOVA,p<0.001),且病毒群落相似性与地理距离多呈负相关(皮尔逊相关系数r<-0.13,p<0.001)。
病毒分类与功能特征

58.3%的vOTUs能与已知基因组匹配,95.9%仅能在纲水平分类(主要为有尾噬菌体纲、逆转录病毒纲、乳头多瘤病毒纲),各城市不同纲病毒相对丰度有差异。将vOTUs与参考病毒基因组聚类,得到697个病毒聚类(VCs),117个VCs可分类,最大且最丰富的VC1(含2430个vOTUs)隶属于花椰菜花叶病毒科,在各城市平均相对丰度46.2±26.4%;第二大VC2(含266个vOTUs)隶属于有尾噬菌体纲未分类科。部分VCs在不同城市相对丰度差异显著,如香港样本VC1丰度显著低于其他城市。
功能注释显示,72.6%的病毒基因无法匹配同源序列,常见功能与DNA结合/调控(如LAGLIDADG核酸内切酶)、转运蛋白(如ABC转运蛋白)相关,裂解、结构和复制功能也较普遍。同时,在159个vOTUs中发现326个独特ARGs,74%与高质量vOTUs相关,主要包括汞抗性基因、糖肽类抗生素抗性基因等,部分ARG亚型(如merP、merR)在香港样本中丰度最高,msrA亚型仅在纽约发现,arnA亚型仅在香港一个vOTU中检测到。
病毒-宿主相互作用与协同进化

约35.8%的vOTUs可关联到体外宿主(分枝杆菌科最常见),20.1%可关联到体内宿主(微球菌科最常见),且不同城市体内宿主存在差异。多数宿主科的宿主丰度与病毒-宿主丰度比(VHR)显著相关,如丹佛、香港等城市肠杆菌科宿主丰度与VHR呈负相关,除丹佛外其他城市微球菌科宿主丰度与VHR呈正相关。

除香港和斯德哥尔摩外,多数城市毒性型vOTUs丰度显著高于温和型(曼-惠特尼检验,p<0.001),且毒性型vOTUs丰度与病毒-微生物丰度比呈正相关(r=0.60-0.85,p<1.0×10⁻⁴),香港、伦敦和奥斯陆温和型vOTUs丰度与该比值呈负相关(r=-0.78至-0.33,p<3.2×10⁻⁵)。

在CRISPR-Acr相互作用方面,从155个rMAGs中提取1988个CRISPR间隔序列,仅52个可与vOTUs关联,53个rMAGs携带CRISPR-Cas系统(以I型为主),部分为城市特有。同时,在79个vOTUs中识别出155个Acr蛋白,可对抗多种CRISPR-Cas系统,且存在城市特异性Acr蛋白,仅8个Acr蛋白在所有城市存在。
此外,在511个vOTUs中识别出1247个假定AMGs,147个可与体内宿主关联,携带AMGs的vOTUs中53.3%为毒性型,31.9%为温和型。常见AMGs功能包括杂项功能、有机氮转化等,编码核糖核苷酸还原酶的AMGs丰度最高,部分AMGs为城市特有,且部分AMGs可能增强宿主适应性,如外膜通道蛋白TolC有助于去除有毒化合物。
讨论
该研究通过分析六个城市公共交通系统空气样本,揭示了建筑环境空气传播病毒的独特特征。研究发现病毒群落存在显著生物地理差异,北美城市病毒多样性较低,可能受气象、环境及交通系统设计影响;大量未分类vOTUs表明建筑环境空气中存在许多新病毒,花椰菜花叶病毒科占主导地位,与其他生态系统常见病毒不同,可能源于多样的微生物来源。
功能上,病毒基因展现出强大的感染、复制和整合能力,且发现多种ARGs,说明空气传播病毒可能是ARGs储存库,存在促进抗生素抗性传播的风险。病毒-宿主相互作用方面,微球菌科是最常见体内宿主,不同城市宿主存在差异,VHR受病毒生活方式影响,在营养匮乏的建筑环境中,毒性型感染是病毒主要生存策略,这与高营养生态系统不同。
CRISPR-Cas系统与Acr蛋白的协同进化有限,可能因毒性病毒占主导、病毒-宿主相遇概率低及数据库局限性导致。而病毒编码的AMGs可能增强宿主对建筑环境的适应性,助力病毒与宿主共存。
不过,研究存在方法学局限性,如病毒识别可能有假阳性/假阴性、样本量和测序深度有待提升、未富集病毒可能影响结果准确性等。未来需优化分析方法、扩大采样范围、开展培养实验,进一步探究病毒生物地理分布、生活方式切换机制及AMGs表达等,以更全面了解建筑环境空气传播病毒。