Introduction
Gene Set Enrichment Analysis (GSEA) 是一种用于分析基因表达数据的计算生物学方法,旨在揭示与特定生物学过程、通路或功能相关的基因表达模式。GSEA 最初由麻省理工学院的研究人员开发 (1),它不同于传统的基因差异分析方法,如 t 检验或ANOVA,这些方法通常关注单个基因的表达差异。 相反,GSEA 专注于整个基因集的表达趋势,这些基因集通常与特定的生物学过程或通路相关联。
在典型的实验中,从属于两类之一的样本集合中生成数千个基因的 mRNA 表达谱,例如,对药物敏感和耐药的肿瘤。 基因可以根据它们在类别之间的差异表达在排序列表L中排序。挑战在于从这个列表中提取含义。 一种常见的方法是关注 L 顶部和底部的少数基因(即那些表现出最大差异的基因),以辨别明显的生物学线索。这种方法有一些主要的局限性:
-
在校正多个假设检验后,没有任何单个基因可以满足统计显著性的阈值,因为相对于微阵列技术固有的噪声,相关的生物学差异是适度(不是很明显)的。
-
或者,人们可能会留下一长串具有统计学意义的基因(太多了),而没有任何统一的生物学主题。解释可能是令人畏惧的和临时的,取决于生物学家的专业领域。
-
单基因分析可能会错过对通路的重要影响。细胞过程常常影响协同作用的基因组。编码代谢途径成员的所有基因增加 20% 可能会显着改变通过该途径的通量,并且可能比单个基因增加 20 倍更重要。
-
当不同的群体研究相同的生物系统时,两项研究中具有统计学意义的基因列表可能显示出令人沮丧的很少的重叠(但是通路可能有重叠)。
以下是 GSEA 的简要介绍和工作原理:
-
基因集定义:首先,GSEA 需要一个事先定义好的基因集合,这些基因通常按照其在特定生物学通路、功能类别或疾病过程中的作用进行组织。这些基因集可以来自公共数据库,如Gene Ontology、KEGG Pathway、Reactome,或者是研究者自己根据文献和领域知识构建的。
-
基因表达数据:GSEA 需要分析的基因表达数据,通常是从微阵列或RNA测序实验中获得的。这些数据包括不同条件或样本中基因的表达水平。
-
排列(permutation)检验:GSEA 的核心思想是通过对基因集的成员在整个基因表达数据中的排列来确定它们的富集程度。具体来说,GSEA 将所有基因根据其在不同条件下的表达水平进行排序。然后,它从基因集的一端开始,计算该基因集中的基因在排序列表中的偏离程度。如果基因集中的基因在排序列表的某个位置中集中排列,说明该基因集富集在特定的生物学过程或通路中。这个过程通过构建一个富集分数(Enrichment Score)来量化。
-
统计显著性:对于每个基因集,GSEA 计算一个富集分数,并基于随机排列检验来估计其统计显著性。如果一个基因集的富集分数在随机排列中的分布表现出显著差异,那么就认为这个基因集在样本中富集。
-
结果可视化:最后,GSEA 会生成结果报告,其中包括富集分数、基因集的统计显著性以及相关通路或功能的信息。这些结果可视化为富集图谱,用于展示不同基因集的富集情况。

总之,GSEA 是一种用于揭示基因表达数据中生物学意义的强大工具,它可以帮助研究人员理解不同生物学过程、通路或功能在不同条件下的活动变化。它已经广泛应用于生物医学研究中,特别是在基因表达分析和疾病机制研究中发挥了重要作用。
Alogrithm
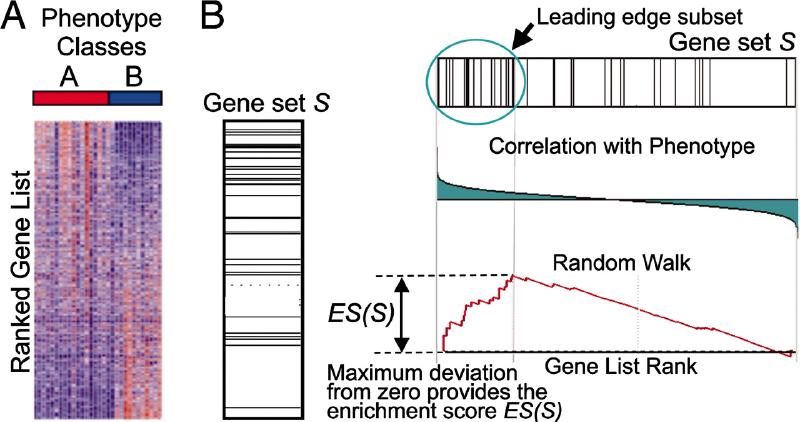
• Step 1: 计算富集分数(Enrichment Score,ES)。计算一个富集分数(ES),反映了一个基因集合S在整个排序列表L的极端位置(顶部或底部)上过度表示的程度。分数的计算是通过遍历列表L来实现的,在遇到基因集S中的基因时,增加一个运行总和统计量;在遇到不在基因集S中的基因时,减少这个统计量。增量的大小取决于基因与表型的相关性。富集分数是在随机漫步中遇到的与零的最大偏差;它对应于一种加权的Kolmogorov–Smirnov-like统计量。
• Step 2: 估计ES的显著性水平。通过使用经验性的基于表型的置换测试过程来估计ES的统计显著性(名义P值),该过程保留了基因表达数据的复杂相关结构。具体来说,对表型标签进行排列并重新计算基因集在排列后数据中的ES,这样生成了ES的零分布。然后,计算观察到的ES的经验性名义P值相对于这个零分布。重要的是,类别标签的排列保留了基因与基因之间的相关性,因此提供了一个更符合生物学的显著性评估,而不是通过排列基因而获得的评估。
实际上GSEA好像提供了两种排列方法 (2),另一种是基因排列,直接将观察到的路径 ES 与通过使用匹配大小(例如 1,000 次)的随机采样基因集重复分析而获得的分数分布进行比较,表型排列应与大量重复一起使用(例如,每个条件至少十次)。 与基因集排列方法相比,表型排列方法的主要优点是,它在排列过程中保持了具有生物学重要基因相关性的基因集结构。 表型排列的计算成本很高,并且对于当前版本的 GSEA,需要自定义编程来分别计算数千个表型随机化的 ES 和差异表达统计数据。
• Step 3: 多重假设检验的调整。当评估整个基因集合数据库时,会调整估计的显著性水平以考虑多重假设检验。首先,对每个基因集的ES进行归一化,以考虑集合的大小,得到一个归一化富集分数(NES)。然后,通过计算与每个NES相对应的虚假发现率(FDR)来控制假阳性比例。FDR是给定NES的一个集合代表虚假阳性发现的估计概率;它是通过比较观察到的NES和零分布的尾部来计算的。
Leading-Edge Subset
基因集可以通过使用多种方法来定义,但并非基因集的所有成员通常都会参与生物过程。通常,提取有助于 ES 的高分基因集的核心成员是有用的。
将Leading-Edge Subset (前沿子集)定义为基因集 S 中出现在排序列表 L 中或之前运行总和达到与零的最大偏差的那些基因(图 1B)。前沿子集可以解释为解释富集信号的基因集的核心。
MSigDB
他们在发表GSEA时同时创建了MSigDB (Molecular Signatures Database),分子特征数据库 (MSigDB) 是一个包含数以万计的注释基因集的资源,可与 GSEA 软件一起使用,分为人类和小鼠集合。
比如说人类分子特征数据库 (MSigDB) 中的 33591 个基因集分为 9 个主要集合和几个子集合,可以自己去下载研究相关的重要基因集:
| Collection | Description |
|---|---|
| H: hallmark gene sets | Hallmark gene sets summarize and represent specific well-defined biological states or processes and display coherent expression. These gene sets were generated by a computational methodology based on identifying overlaps between gene sets in other MSigDB collections and retaining genes that display coordinate expression. details |
| C1: positional gene sets | Gene sets corresponding to human chromosome cytogenetic bands. details |
| C2: curated gene sets | Gene sets in this collection are curated from various sources, including online pathway databases and the biomedical literature. Many sets are also contributed by individual domain experts. The gene set page for each gene set lists its source. The C2 collection is divided into the following two subcollections: Chemical and genetic perturbations (CGP) and Canonical pathways (CP). details |
| C3: regulatory target gene sets | Gene sets representing potential targets of regulation by transcription factors or microRNAs. The sets consist of genes grouped by elements they share in their non-protein coding regions. The elements represent known or likely cis-regulatory elements in promoters and 3’-UTRs. The C3 collection is divided into two subcollections: microRNA targets (MIR) and transcription factor targets (TFT). details |
| C4: computational gene sets | Computational gene sets defined by mining large collections of cancer-oriented microarray data. The C4 collection is divided into two subcollections: CGN and CM. details |
| C5: ontology gene sets | Gene sets that contain genes annotated by the same ontology term. The C5 collection is divided into two subcollections, the first derived from the Gene Ontology resource (GO) which contains BP, CC, and MF components and a second derived from the Human Phenotype Ontology (HPO). details |
| C6: oncogenic signature gene sets | Gene sets that represent signatures of cellular pathways which are often dis-regulated in cancer. The majority of signatures were generated directly from microarray data from NCBI GEO or from internal unpublished profiling experiments involving perturbation of known cancer genes. details |
| C7: immunologic signature gene sets | Gene sets that represent cell states and perturbations within the immune system. details |
| C8: cell type signature gene sets | Gene sets that contain curated cluster markers for cell types identified in single-cell sequencing studies of human tissue. details |
Table 1: Gene sets in the Human Molecular Signatures Database .
Example
这里介绍在R中完成GSEA的方法
- 对GSEA分析的geneList排序
明确我们用来排序的指标(metric)。 目前大部分分析是只比较两组间差异的,所以会将基因按照在两类样本中的差异表达程度(一般是log2FoldChange)排序,但实际上这里可以使用多种指标比如相关性,p值,也可以直接进行多组的分析比较(当然一定要明确排序两端代表的生物学意义)。尽量不要进行原始基因表格的筛选,比如只取DEGs,因为GSEA本身就是一个无阈值的富集方法,相比fisher.test等阈值方法考虑的会更多一些。
|
|
## 4312 8318 10874 55143 55388 991
## 4.572613 4.514594 4.418218 4.144075 3.876258 3.677857
- 使用R包进行GSEA富集
如果要使用GSEA官网提供的带GUI软件,可以参考这篇nature protocol,里面详细介绍了每一步的操作 (2)。
这里我们使用更方便一点的R包来进行。clusterProfiler包内的gseGO()和gseKEGG()函数可以很方便地对GO与KEGG通路进行GSEA。 如果明确我们要富集的基因集的话,也可以自己构建并使用clusterProfiler::GSEA()富集。
|
|
但上面这行代码在我的机器上结果是– No gene can be mapped….,可能是我机器上的clusterProfiler包内部数据库问题。
但是没有关系,我一般也会自己从kegg上把想分析的通路信息都爬下来,操作见上一篇关于kegg api的介绍, 具体的代码都集成在ReporterScore里面了,下面直接使用ReporterScore进行GSEA分析。
|
|
## pathway_id kegg_gene_id gene_symbol gene_desc KO_id
## 1 hsa00010 3101 HK3 hexokinase 3 K00844
## 2 hsa00010 3098 HK1 hexokinase 1 K00844
## 3 hsa00010 3099 HK2 hexokinase 2 K00844
## 4 hsa00010 80201 HKDC1 hexokinase domain containing 1 K00844
## 5 hsa00010 2645 GCK glucokinase K12407
## 6 hsa00010 2821 GPI glucose-6-phosphate isomerase K01810
|
|
## KO_id average_WT sd_WT average_OE sd_OE
## MME MME 0.0026728975 0.0011094132 0.0020694937 0.0014922004
## PEX11A PEX11A 0.0004554658 0.0001951678 0.0024480601 0.0014916566
## DSC2 DSC2 0.0027767713 0.0006253559 0.0025519275 0.0004966801
## PLIN5 PLIN5 0.0005779169 0.0008952163 0.0005197504 0.0001435245
## MIRLET7A2 MIRLET7A2 0.0020807307 0.0007731661 0.0014321838 0.0004273716
## NCBP2L NCBP2L 0.0021064422 0.0005243558 0.0017419317 0.0005382770
## diff_mean Highest p.value p.adjust
## MME -0.00060340387 WT 0.2200529827 0.321714887
## PEX11A 0.00199259427 OE 0.0001376795 0.001449258
## DSC2 -0.00022484380 WT 0.2852910552 0.392962886
## PLIN5 -0.00005816647 WT 0.8072016928 0.865027977
## MIRLET7A2 -0.00064854693 WT 0.0095033556 0.028624565
## NCBP2L -0.00036451047 WT 0.0707452991 0.134260284
|
|
## ID
## hsa05235 hsa05235
## hsa05162 hsa05162
## hsa05323 hsa05323
## hsa05322 hsa05322
## hsa04660 hsa04660
## hsa04142 hsa04142
## Description
## hsa05235 PD-L1 expression and PD-1 checkpoint pathway in cancer - Homo sapiens (human)
## hsa05162 Measles - Homo sapiens (human)
## hsa05323 Rheumatoid arthritis - Homo sapiens (human)
## hsa05322 Systemic lupus erythematosus - Homo sapiens (human)
## hsa04660 T cell receptor signaling pathway - Homo sapiens (human)
## hsa04142 Lysosome - Homo sapiens (human)
## setSize enrichmentScore NES pvalue p.adjust qvalue rank
## hsa05235 13 0.8015443 1.885221 0.005680812 0.3204232 0.3178287 118
## hsa05162 18 0.7355862 1.857939 0.006161984 0.3204232 0.3178287 14
## hsa05323 14 0.7751069 1.850851 0.005438948 0.3204232 0.3178287 1
## hsa05322 15 0.7364826 1.787932 0.011670053 0.3798506 0.3767749 101
## hsa04660 15 0.7348533 1.783977 0.012174699 0.3798506 0.3767749 43
## hsa04142 20 -0.5462876 -1.628928 0.018884445 0.4909956 0.4870199 40
## leading_edge core_enrichment
## hsa05235 tags=46%, list=12%, signal=41% CD28/TICAM1/MAPK13/ALK/RPS6KB2/TLR9
## hsa05162 tags=11%, list=1%, signal=11% CD28/TAB2
## hsa05323 tags=7%, list=0%, signal=7% CD28
## hsa05322 tags=27%, list=10%, signal=24% CD28/H4C13/H3C15/H2BW2
## hsa04660 tags=13%, list=4%, signal=13% CD28/MAPK13
## hsa04142 tags=15%, list=4%, signal=15% AP1M2/SLC17A5/SLC11A2
-
ID:某条通路基因集的名字
-
Description:通路的文字描述
-
setSize:gene set(S)中的基因数目(经过条件筛选后的值)
-
enrichmentScore:富集评分
-
NES:校正后的归一化的ES值
-
pvalue:对富集得分ES的统计学分析,用来表征富集结果的可信度
-
p.adjust:对pvalue进行BH调整后的值
-
qvalue:即多重假设检验校正之后的pvalue。对NES可能存在的假阳性结果的概率估计,因此FDR越小说明富集越显著;
-
rank:取到ES值时,对应基因在排序好的基因列表中所处的位置
-
leading_edge:该处有3个统计值
-
ES>0在左边,ES<0在右边
-
tags=xx%表示peak gene在S中的位置。指示有贡献的gene数量。
-
list=xx%表示peak gene在L中的位置。指示ES在哪里得到。
-
signal计算:$(Tag%)(1-Gene%)(\frac{N}{N-Nh})$
-
N:L中的gene数量
-
Nh:S中的gene数量
-
-
- 结果可视化 使用enrichplot包对富集结果进行可视化。
|
|
|
|
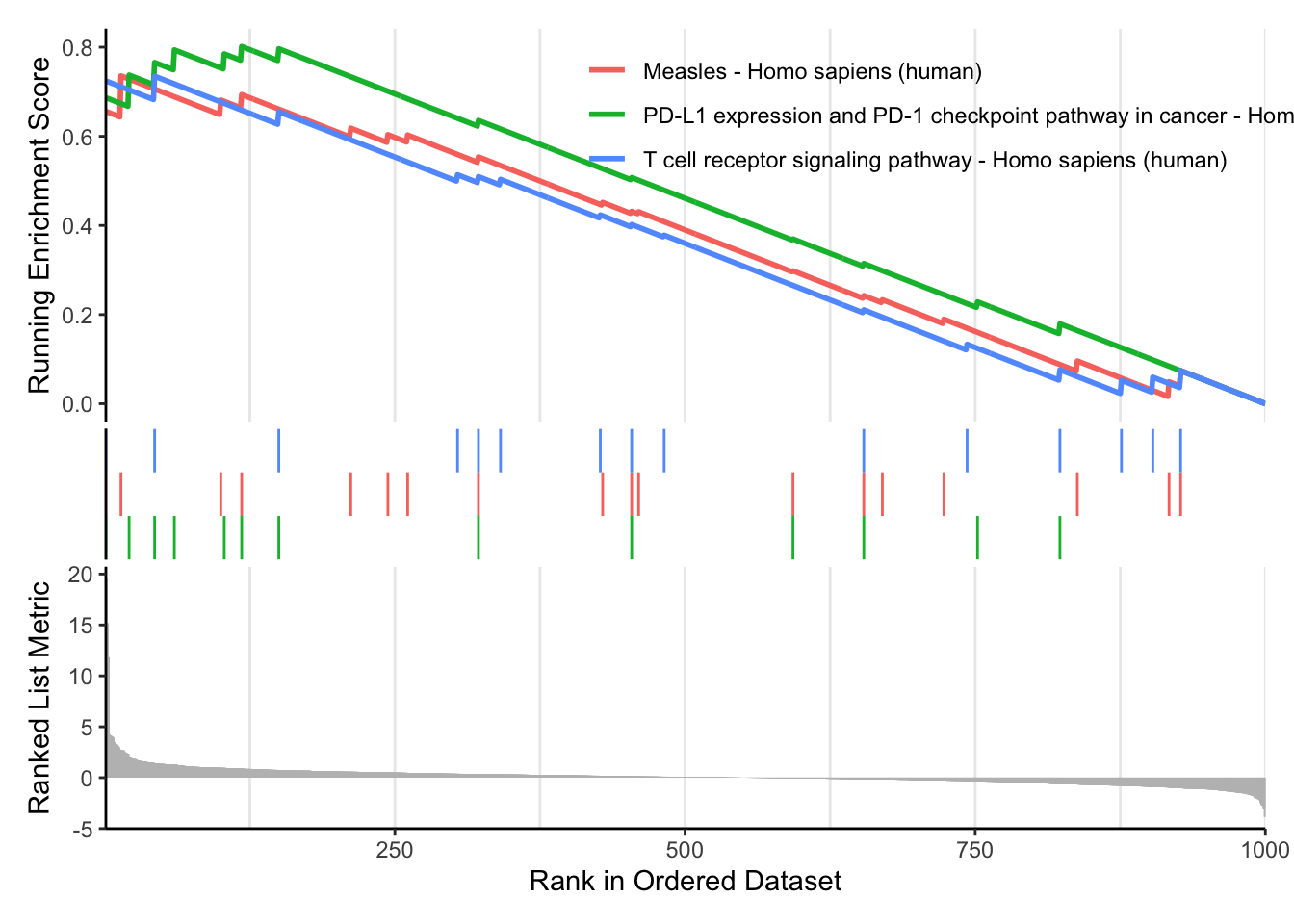
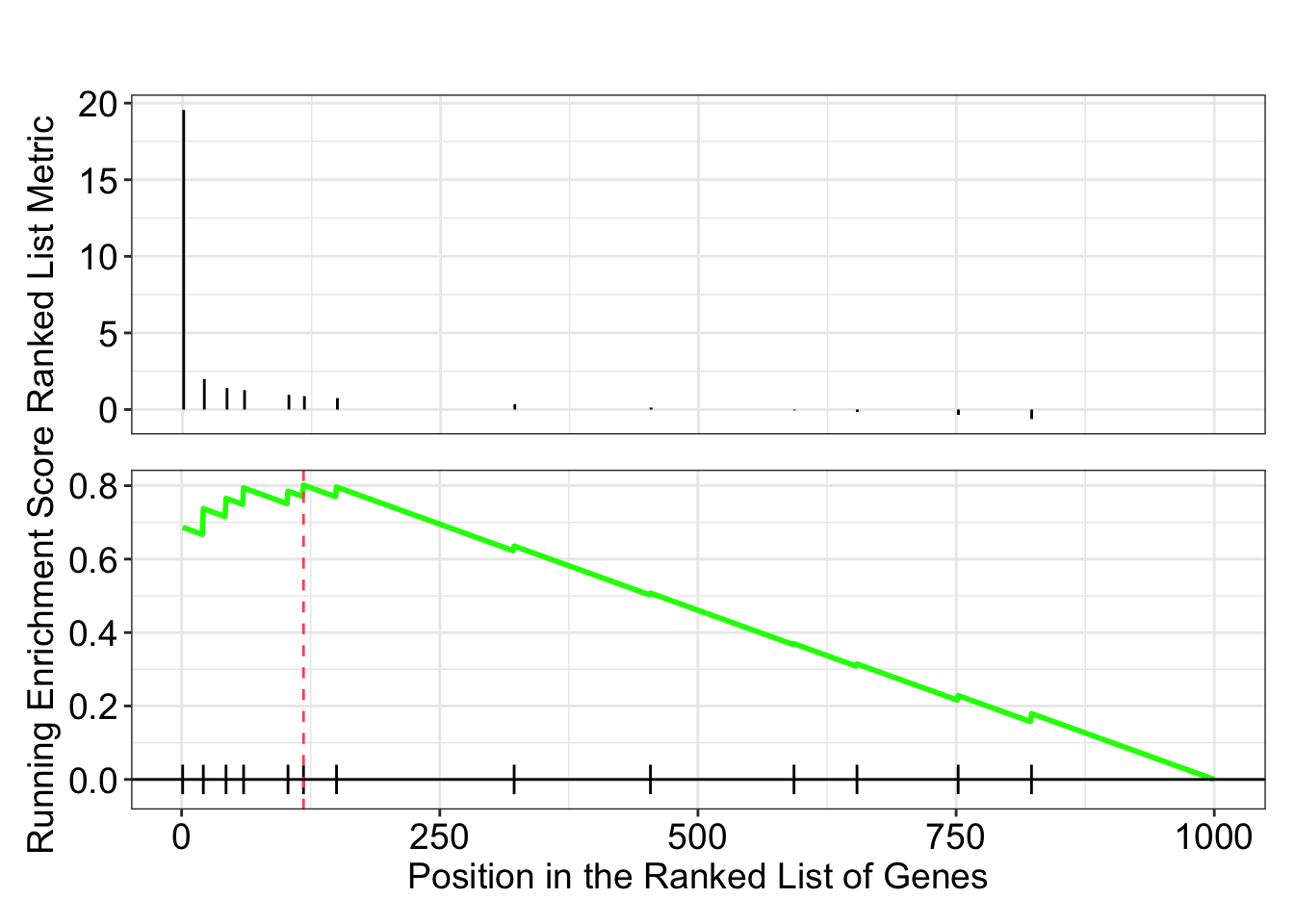
第1部分 - ES折线图:在ES折线图中,离垂直距离x=0轴最远的峰值便是基因集的ES值。峰出现在排序基因集的前端(ES值大于0)则说明通路上调,出现在后端(ES值小于0)则说明通路下调。
第2部分 - 基因集成员位置图:在该图中,用竖线标记了基因集中各成员出现在基因排序列表中的位置。若竖线集中分布在基因排序列表的前端或后端,说明该基因集通路上调或下调;若竖线较均匀分布在基因排序列表中,则说明该基因集通路在比较的两个数据中无明显变化。 红色部分对应的基因在实验组中高表达,蓝色部分对应的基因在对照组中高表达, leading edge subset 是(0,0)到曲线峰值ES出现对应的这部分基因成员。
第3部分 - 排序后所有基因rank值分布:该图展示了排序后的所有基因rank值(由log2FoldChange值计算得出)的分布,以灰色面积图显展示。
Reference
links:
https://zhuanlan.zhihu.com/p/581172803
https://zhuanlan.zhihu.com/p/518144716
1. A. Subramanian, P. Tamayo, V. K. Mootha, S. Mukherjee, et al., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences. 102, 15545–15550 (2005).
2. J. Reimand, R. Isserlin, V. Voisin, M. Kucera, et al., Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, cytoscape and EnrichmentMap. Nature Protocols. 14, 482–517 (2019).