Introduction
序列比对是生物信息学研究的基础操作,从基因功能注释、病原体溯源到进化分析,都离不开将查询序列与参考基因组数据库进行比对。然而,随着高通量测序技术的飞速发展,公共数据库中的原核生物基因组数量呈指数级增长——GenBank与RefSeq联合数据库已包含超过234万个原核生物基因组,AllTheBacteria高质量数据集也收录了185万余个细菌基因组。
传统序列比对工具在此背景下面临严峻挑战:BLASTn虽准确性高,但对百万级基因组数据库需超过2000GB内存,普通服务器根本无法运行;MMseqs2、Minimap2等工具虽在速度上有提升,却难以平衡内存消耗与结果完整性;基于“预过滤+比对”的策略(如Phylign)则在序列分歧度较高时出现敏感性下降。
《Nature Biotechnology》发表的研究 “Efficient sequence alignment against millions of prokaryotic genomes with LexicMap” 提出了突破性解决方案。研究者开发的LexicMap工具,通过创新的种子设计与索引策略,实现了对百万级原核生物基因组的快速、低内存比对,同时保持与主流工具相当的准确性,为大规模基因组分析提供了新选择。
这里简单介绍一下这篇文章,并提供LexicMap工具的使用教程。
文章介绍
发表于《Nature Biotechnology》的这项研究,针对大规模基因组比对的核心痛点,设计了一套兼顾速度、内存与准确性的解决方案。研究团队通过优化序列草图生成、种子匹配与索引构建流程,开发出LexicMap工具,并在多个百万级基因组数据集上验证了其性能优势。
序列比对工具面临的规模化挑战
当前主流序列比对工具在处理大规模基因组数据时,主要存在三大瓶颈:
- 内存消耗过高:传统工具(如BLASTn、Ropebwt3)对100万个基因组建库时,内存需求可达数百甚至上千GB,远超普通计算设备的承载能力;
- 速度与敏感性难以平衡:基于k-mer的比对工具(如COBS)虽速度快,但仅能返回匹配基因组,无法提供关键的位置信息;而“预过滤+比对”工具(如Phylign)在序列分歧度超过10%时,预过滤步骤会大量丢失有效匹配;
- 结果完整性不足:部分映射工具(如Minimap2)仅返回相似度最高的匹配,忽略了基因组中多拷贝基因或片段化匹配,影响下游分析的全面性。
为解决这些问题,LexicMap从算法底层进行创新,核心目标是在“大规模”,“高准确性”,“低资源消耗”三者间找到最优平衡。
LexicMap的核心设计与创新
LexicMap的优势源于四项关键技术创新,形成了从“种子生成”到“结果输出”的完整优化流程:

1. 基于LexicHash的精准种子生成
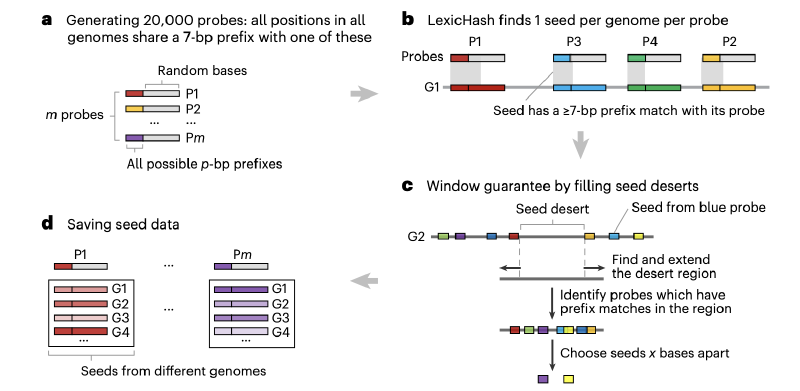
LexicMap改进了序列草图工具LexicHash,通过生成2万个31-mer“探针”(probe)覆盖所有可能的7-mer前缀——这意味着数据库中每个基因组的任意250-bp窗口,都会包含至少2个(实际中位数为5个)与探针前缀匹配的k-mer(称为“种子”)。这种设计确保了种子在基因组上的均匀分布,避免了传统工具中“种子荒漠”(seed desert)导致的比对缺失问题。
2. 前缀+后缀双匹配策略
为提升对序列突变的耐受性,LexicMap不仅支持种子的前缀匹配,还新增了后缀匹配功能:将种子反向互补后,与探针进行二次前缀匹配,相当于实现了后缀匹配。这种双匹配策略使工具能有效识别包含插入、缺失或碱基替换的序列,尤其在序列分歧度较高(如80%-90%)时,敏感性显著优于固定长度k-mer匹配工具。

3. 分层索引与批量建库
针对百万级基因组的存储与查询需求,LexicMap采用“批量建库+分层索引”策略:
- 批量建库:将基因组按5000-25000个为一批次分别建库,每批内将基因组的多条contig用1kb N序列连接,减少索引复杂度,最后合并所有批次索引,大幅降低单批次内存消耗;
- 分层索引:种子数据按探针分组存储,每组内通过varint-GB算法压缩,同时建立“目录式”索引(类似字典目录),查询时可快速定位种子位置,避免全库扫描。
4. 伪比对+Wavefront精确比对
LexicMap采用“两步比对”流程保证结果准确性:
- 伪比对(Pseudoalignment):对种子匹配后的区域进行扩展,通过2-mer匹配快速筛选出高相似度区域,减少后续精确比对的计算量;
- Wavefront精确比对:使用Wavefront算法进行碱基级比对,计算并输出与BLASTn一致的关键指标(如qcovGnm、pident、evalue、bitscore),确保结果的可解释性与兼容性。
LexicMap的性能优势与准确性验证
研究团队在三个大规模数据集(GTDB r214:40万个原核基因组;AllTheBacteria:185万个细菌基因组;GenBank+RefSeq:234万个原核基因组)上,将LexicMap与BLASTn、MMseqs2、Minimap2、Ropebwt3等工具进行全面对比,结果显示其在“速度”“内存”“准确性”上均表现突出。


1. 规模化比对的速度与内存优势
在100万个基因组的数据库中,LexicMap的优势尤为显著:
- 索引构建:内存消耗仅75GB,远低于Ropebwt3(1013GB)、COBS(382GB),虽略高于MMseqs2(20GB),但索引大小仅为Minimap2的1/8;
- 查询速度:对1.3kb标记基因(secY)的比对仅需3分06秒,是MMseqs2(89倍慢)、Minimap2(39倍慢)的数十倍,且内存消耗仅3.97GB,而BLASTn在此规模下无法运行;
- 长期稳定性:在234万个基因组的GenBank+RefSeq数据集上,LexicMap可稳定运行,单查询内存不超过15GB,而其他工具要么内存溢出,要么查询时间超过数小时。
2. 准确性与敏感性验证
通过模拟含不同突变率(0%-20%)的序列(长度250bp-2000bp)进行验证,LexicMap的准确性表现如下:
- 高相似度序列(≥95%):所有工具的比对率均接近100%,LexicMap与BLASTn(word size=15)、MMseqs2的结果完全一致;
- 中低相似度序列(80%-93%):对于250bp短序列,LexicMap在88%相似度以下的比对率超过Minimap2,在93%相似度以下超过默认参数的BLASTn(word size=28);
- 实际数据验证:对53kb质粒、16S rRNA基因及1033个抗生素抗性基因(AMR)的比对显示,LexicMap能准确识别多拷贝基因(如基因组中的多个16S rRNA拷贝),且低相似度匹配(<80%)的假阳性率低于BLASTn和MMseqs2。
LexicMap在生物学研究中的应用价值
LexicMap的设计充分考虑了实际研究需求,可广泛应用于以下场景:
- 病原体溯源与流行病学分析:快速比对临床样本中的耐药基因或质粒,定位其在全球基因组数据库中的分布,追踪传播路径(如霍乱弧菌耐药质粒的跨地区传播);
- 微生物生态学研究:对环境样本中的长读长序列(如Nanopore reads)进行比对,识别低丰度物种或新型菌株,避免传统工具因敏感性不足导致的物种遗漏;
- 功能基因挖掘:批量比对未注释基因组中的功能基因(如代谢酶基因),通过比对结果预测基因功能,尤其适用于未被收录在参考数据库中的新基因;
- 进化分析:通过比对不同物种的同源基因,计算序列分歧度,构建进化树,LexicMap的双匹配策略可有效处理进化距离较远的序列。
研究还特别指出,LexicMap返回的“基因组级查询覆盖率”(qcovGnm)指标,对环状序列(如质粒、病毒基因组)的比对结果解读至关重要——可区分完整匹配与片段化匹配,避免将环状序列的线性比对误判为不完整匹配。
LexicMap 使用教程
LexicMap提供Linux、Windows、MacOS(x86/arm架构)的预编译二进制文件,无需依赖其他软件,安装与使用流程简洁。同时支持在线比对(部分功能)与本地部署,满足不同规模的分析需求。
1. 安装准备
环境要求
- 操作系统:Linux(CentOS 7+/Ubuntu 18+)、Windows 10+、MacOS 12+;
- 硬件配置:最低8GB内存(适用于10万个基因组以下数据库),建议32GB以上内存(百万级基因组数据库);
- 存储:根据数据库规模,需预留100GB-10TB空间(如234万个基因组的索引约5.46TB),推荐使用SSD以提升查询速度。
安装方式
LexicMap支持三种安装方式,推荐使用二进制文件或conda安装,步骤如下:
方式1:二进制文件安装
- 下载对应系统的二进制文件:访问LexicMap GitHub Release页面,下载最新版本(如v0.7.0)的压缩包(例:
lexicmap_v0.7.0_linux_amd64.tar.gz); - 解压并配置环境变量:
1 2 3 4 5 6# 解压 tar -zxvf lexicmap_v0.7.0_linux_amd64.tar.gz # 移动到可执行目录(如/usr/local/bin) sudo mv lexicmap /usr/local/bin/ # 验证安装 lexicmap --version
方式2:conda安装
通过bioconda频道快速安装,适用于已配置conda环境的用户:
|
|
方式3:源码编译
需安装Go 1.20+环境,适合需要自定义功能的用户:
|
|
2. 输入文件与数据要求
LexicMap支持多种输入格式,核心是“基因组数据库”与“查询序列”,具体要求如下:
基因组数据库输入
- 格式:FASTA格式(支持.gz、.xz、.zstd等压缩格式),每个基因组单独一个FASTA文件,或多个基因组合并为一个FASTA文件(需确保序列ID唯一);
- 命名规则:文件名建议包含基因组ID(如
GCF_000005845.2.fasta),便于后续结果关联; - 输入方式:可通过目录(
-I参数)、文件列表(-X参数,每行一个FASTA文件路径)或单个文件(-i参数)指定。
查询序列输入
- 格式:FASTA格式(支持压缩),单个或多个查询序列(如基因、质粒、长读长);
- 长度要求:推荐≥250bp,最短支持100bp(短于100bp时敏感性可能下降);
- 特殊类型:支持环状序列(如质粒、病毒),LexicMap会通过
qcovGnm指标识别完整匹配。
输出结果格式
LexicMap默认输出制表符分隔的TSV文件,包含以下关键列(与BLASTn兼容,便于后续分析):
| 列名 | 含义 | 示例值 |
|---|---|---|
| query | 查询序列ID | secY_Efaecalis |
| qlen | 查询序列长度 | 1299 |
| sgenome | 匹配的基因组ID | GCF_000392875.1 |
| sseqid | 匹配的基因组序列ID(如contig) | NZ_KB944588.1 |
| qcovGnm | 查询序列在基因组中的整体覆盖率 | 100.000 |
| pident | 序列相似度(百分比) | 98.701 |
| evalue | 期望值(越小越可靠) | 1.93e-63 |
| bitscore | 比特值(越大越可靠) | 253 |
| sstart/send | 匹配在基因组中的起始/终止位置 | 228637/229935 |
| sstr | 匹配链方向(+:正链,-:负链) | + |
此外,可通过-a/--all参数输出CIGAR字符串、比对序列等详细信息,或通过--format blast输出BLAST风格的 pairwise 比对结果。
3. 核心功能使用(建库与比对)
LexicMap的核心命令是index(构建数据库索引)与search(执行序列比对),以下为完整流程示例。
步骤1:构建基因组数据库索引
以“将AllTheBacteria数据集的1万个基因组构建索引”为例,命令如下:
|
|
- 关键参数说明:
--batch-size:批次大小影响内存消耗,内存充足时可增大(如25000),内存有限时减小(如1000);--force:强制覆盖已存在的索引目录;--resume:恢复中断的建库过程(适用于大规模建库)。
建库完成后,索引目录allthebacteria_10k.lmi包含以下文件:
index.meta:索引元信息(如基因组数量、探针数量);seed.chunk.*:种子数据文件(按探针分组);genome.info:基因组信息(长度、序列ID等)。
步骤2:执行序列比对
以“查询1个16S rRNA基因(rrsB_Ecoli.fasta)”与“批量查询1033个AMR基因(amr_genes.fasta)”为例,展示两种常见场景的使用方法。
场景1:单查询序列比对(16S rRNA基因)
|
|
场景2:批量查询序列比对(AMR基因)
批量查询时,可通过--split-query参数将查询序列拆分到多个线程,提升速度:
|
|
步骤3:结果可视化与二次分析
LexicMap提供实用工具命令lexicmap utils,支持结果筛选、序列提取与格式转换,示例如下:
提取高相似度匹配的基因组序列
|
|
转换为BLAST风格的 pairwise 比对格式
|
|
4. 常见问题与优化建议
问题1:建库时内存不足
- 解决方案:减小
--batch-size参数(如从5000改为1000),或使用--tmp-dir指定临时目录(避免内存交换); - 示例:
lexicmap index -I ./genomes -O ./index --batch-size 1000 --tmp-dir /mnt/tmp。
问题2:查询速度慢
- 优化方向:
- 使用SSD存储索引目录(随机读取速度提升10-100倍);
- 对非必需的低相似度匹配进行过滤(如
--min-pident 85、--evalue 1e-20); - 批量查询时增大
--split-query参数(如设为线程数的2倍)。
问题3:未找到预期匹配
- 排查步骤:
- 检查查询序列格式是否正确(FASTA头是否完整,有无特殊字符);
- 降低过滤阈值(如
--min-pident 80、--min-qcov-per-genome 50); - 使用
lexicmap utils index-info查看索引是否包含目标基因组:1lexicmap utils index-info -d ./index | grep "目标基因组ID"
References
- Shen, W., Lees, J.A. & Iqbal, Z. (2025). Efficient sequence alignment against millions of prokaryotic genomes with LexicMap. Nature Biotechnology. https://doi.org/10.1038/s41587-025-02812-8
- LexicMap 官方文档:https://bioinf.shenwei.me/LexicMap/introduction/
- LexicMap GitHub 仓库:https://github.com/shenwei356/LexicMap
- AllTheBacteria 数据集:https://github.com/AllTheBacteria/AllTheBacteria
- GTDB 数据库:https://gtdb.ecogenomic.org/