Introduction
微生物组研究的一个核心问题是识别与特定表型(如健康状况或环境条件)相关的微生物特征。然而,微生物组数据具有稀疏性、组成性和异方差性等特点,使得统计分析面临巨大挑战。近期发表在bioRxiv上的论文《MaAsLin 3: Refining and extending generalized multivariable linear models for meta-omic association discovery》提出了一个强大的解决方案。
MaAsLin 3文章简介
MaAsLin3 (Microbiome Multivariable Associations with Linear Models) 是哈佛大学Huttenhower C.大佬团队开发的微生物组多变量关联分析R包工具,相比前代主要有三大突破:
- 同时检测丰度和存在性关联
- 传统方法只能检测微生物相对丰度的变化
- MaAsLin 3通过分步建模,分别检测:
- 存在/缺失(Prevalence)
- 非零时的丰度变化(Abundance)

-
创新的组成性校正方法
- 通过中位数系数比较策略推断绝对丰度变化
- 支持实验性绝对定量数据(如spike-in)
-
扩展的分析能力
- 支持混合效应模型
- 新增有序预测变量分析
- 支持特征特异性协变量(如宏转录组中的DNA丰度)
性能表现
在模拟数据测试中,MaAsLin 3展现出显著优势:
- 在50个以上样本时F1分数最高
- 平均精确度≥0.82,优于同类工具
- 系数估计偏差最小(仅-12%)

在IBD研究中的新发现
应用MaAsLin 3分析HMP2炎症性肠病(IBD)数据库时发现:
- 77%的显著关联是存在性(而非丰度)变化
- 确认了成人IBD中Enterocloster spp.的富集
- 首次发现Dysosmobacter welbionis在IBD中的存在性降低

实际应用建议
-
数据类型选择:
- 优先使用spike-in或qPCR绝对定量数据
- 相对丰度数据需启用中位数校正
-
模型设定:
1 2 3 4 5 6 7 8# 基础模型 maaslin3( input_data = features, input_metadata = metadata, normalization = "TSS", transform = "LOG", analysis_method = "LM" ) -
结果解读:
- 关注q-value<0.1且|β|>1的关联
- 区分丰度与存在性变化的生物学意义
使用教程
官方教程:https://github.com/biobakery/biobakery/wiki/MaAsLin3
安装MaAsLin 3
最新开发版的MaAsLin 3可通过devtools工具包从GitHub安装。
稳定版MaAsLin 3可通过BiocManager安装:
|
|
运行前需加载依赖库:
|
|
使用MaAsLin 3进行微生物组关联分析
运行MaAsLin 3需要提供以下数据:
- 样本特征丰度表(需保留零值)
- 样本元数据表
- 指定元数据与特征存在率(特征出现概率)及丰度(存在时的数量级)关系的公式或固定效应(可添加随机效应)
输出结果包括:
- 关联分析表(含每个特征-元数据组合的效应量及p值)
- 可视化文件夹(含显著关联的摘要图及诊断图)
输入文件要求
需准备两个输入文件:
1.特征丰度数据框
- 格式1:列=特征,行=样本
- 格式2:行=特征,列=样本(转置格式亦可)
- 特征类型:分类单元或基因(支持相对丰度或绝对计数)
- 可接受制表符分隔的文件路径
2.元数据数据框
- 格式1:列=变量,行=样本
- 格式2:行=变量,列=样本(转置格式亦可)
- 变量类型:性别、年龄等
- 可接受制表符分隔的文件路径
注意事项:
- 允许存在仅出现在一个文件中的样本,分析时会自动剔除不匹配样本
- 两个文件的样本顺序无需一致,程序会自动对齐
- 含NA值的样本在模型拟合时会被排除,建议在随机缺失假设下预先剔除或使用多重插补处理
示例文件说明: 示例数据来自人类微生物组计划2(HMP2),存放于MaAsLin 3源码的inst/extdata目录或教程文件库:
- HMP2_taxonomy.tsv:物种丰度子集(行=样本,列=物种)
- HMP2_metadata.tsv:元数据子集(行=样本,列=变量)
数据读取与预处理:
|
|
|
|
|
|
|
|
运行MaAsLin 3分析
MaAsLin 3的运行需要指定以下参数:
- 丰度表(
input_data) - 元数据表(
input_metadata) - 输出目录(
output) - 分析模型(可通过公式或变量向量指定)
模型构建方式
-
公式法:
- 遵循lme4语法规范,可包含:
- 固定效应
- 随机效应
- 交互项
- 多项式项
- 分类变量作为固定效应时,各水平将与首个因子水平对比
- 支持特殊声明:
group(variable_name):分组预测变量ordered(variable_name):有序预测变量strata(variable_name):配对样本的条件逻辑回归
- 遵循lme4语法规范,可包含:
-
向量法:
- 通过以下参数指定变量:
fixed_effects:固定效应random_effects:随机效应group_effects:分组变量ordered_effects:有序变量strata_effects:配对变量
- 注意事项:
- 需使用列名而非
$符号引用变量 - 分类变量需预先因子化,或通过
'变量,参考水平;...'格式指定(如diagnosis,nonIBD;antibiotics,No)
- 需使用列名而非
- 通过以下参数指定变量:
重要建议
- 当存在测序深度数据时,应将其作为协变量纳入模型(
reads参数),以避免因测序深度差异导致的假阳性关联
HMP2数据实战示例
以下代码演示如何分析微生物物种与IBD诊断的关联,同时控制抗生素使用、年龄和测序深度的影响:
|
|
关键参数解析
-
标准化与转换
TSS+LOG组合是推荐配置,其他选项详见?maaslin3
-
统计检验策略
- 丰度关联:默认与特征的中位数系数比较(
median_comparison_abundance=TRUE),适用于相对丰度数据 - 存在率关联:默认与0比较(
median_comparison_prevalence=FALSE)
- 丰度关联:默认与特征的中位数系数比较(
-
适用场景建议
- 关闭中位数比较的情况:
- 研究相对丰度关联
- 假设总绝对丰度不变时的绝对关联分析
- 使用spike-in或未标准化丰度数据时
- 关闭中位数比较的情况:
-
输出控制
- 默认输出详细日志,可通过
verbosity = 'WARN'减少输出
- 默认输出详细日志,可通过
中位数比较机制详解
当启用median_comparison时:
- 丰度系数:与所有特征在该元数据上的中位数系数对比,抵消组成性效应
- 仅影响p值/q值计算(除非设置
subtract_median=TRUE才会修正系数值)
- 仅影响p值/q值计算(除非设置
- 存在率系数:通常保持与0比较,因其不受组成性影响
该设计确保了:
- 相对丰度数据可推断绝对丰度变化
- 避免因技术偏差(如测序深度)导致的假关联
3.3 MaAsLin 3 输出结果解析
MaAsLin 3的输出包含数据文件和可视化图表两类,完整示例可参考教程文件库中的HMP2分析结果。
核心结果文件
1. 显著性关联表 (significant_results.tsv)
按q值升序列出所有通过显著性检验的关联,字段说明:
| 字段 | 说明 | 示例值 |
|---|---|---|
feature |
微生物特征名 | Phocaeicola_sartorii |
metadata |
元数据变量名 | reads |
coef |
效应量系数 | 1.095 |
| - 丰度模型:表示元数据每增加1单位,特征相对丰度的2^coef倍变化 | ||
| - 存在率模型:表示对数几率(log-odds)的变化量 | ||
null_hypothesis |
零假设值(0或中位数) | 0 |
qval_individual |
个体检验FDR校正q值 | 1.06E-40 |
qval_joint |
联合检验q值(基于Beta(1,2)分布计算) | 1.14E-40 |
model |
模型类型(abundance/prevalence) | prevalence |
完整输出文件结构
数据文件
| 文件/目录 | 内容 |
|---|---|
all_results.tsv |
全量关联结果(含错误信息) |
features/ |
处理后的特征表(过滤→标准化→转换) |
models_*.rds |
模型对象(需save_models=TRUE) |
residuals_*.rds |
模型残差(线性模型为普通残差,逻辑模型为偏差残差) |
maaslin3.log |
完整运行日志(含参数设置与报错信息) |
可视化文件
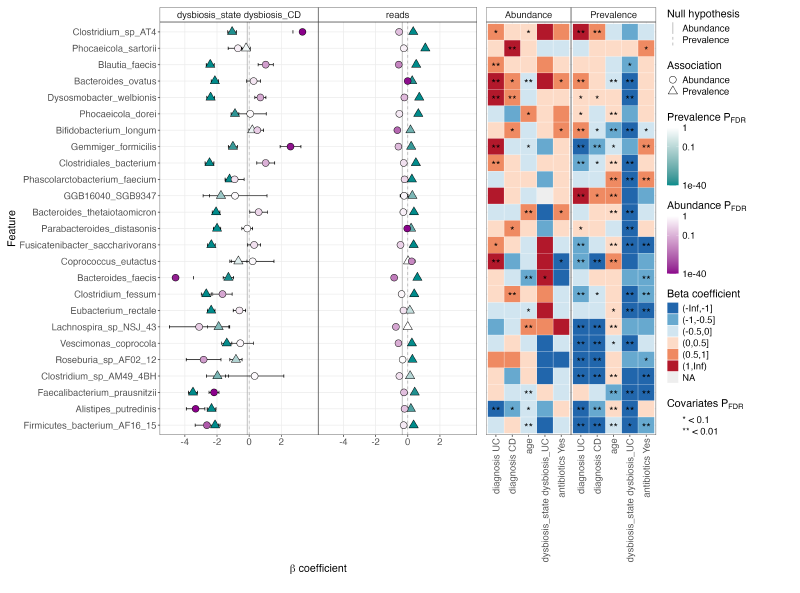
- 摘要图 (
summary_plot.pdf)- 左:系数热图(*表示q<0.1,**表示q<0.01)
- 右:显著关联的效应量排序图

- 关联详图 (
association_plots/)- 连续变量:散点图(丰度)或箱线图(存在率)
- 分类变量:箱线图(丰度)或网格图(存在率)
- 图表右上角标注:q值、总样本数、非零样本数

诊断与验证要点
-
错误排查
- 检查
all_results.tsv的error列:模型拟合失败会标记错误 - 启用
warn_prevalence=TRUE时,存在率关联可能受丰度影响(需通过可视化验证)
- 检查
-
有效性验证
- 分类变量:每组至少10个样本(丰度模型)且存在/缺失样本均≥10(存在率模型)
- 连续变量:通过散点图检查异常值影响
- 超大系数(|coef|>15):需验证:
- 样本量充足(建议样本数≥10×变量数)
- 元数据无多重共线性
- 随机效应设置合理
-
多重检验校正
- 默认对所有关联进行FDR校正,若需聚焦特定变量,可单独校正目标p值
结果重绘功能
通过maaslin_plot_results_from_output可重新生成图表(无需原始R对象):
|
|
更多高级功能,请查看https://github.com/biobakery/biobakery/wiki/MaAsLin3#4-advanced-topics
References
- Nickols, W.A., Kuntz, T., Shen, J., Maharjan, S., Mallick, H., Franzosa, E.A., Thompson, K.N., Nearing, J.T., and Huttenhower, C. (2024). MaAsLin 3: refining and extending generalized multivariable linear models for meta-omic association discovery. Preprint at bioRxiv, https://doi.org/10.1101/2024.12.13.628459 https://doi.org/10.1101/2024.12.13.628459.
- https://github.com/biobakery/biobakery/wiki/MaAsLin3