Introduction
Panaroo是一个用于微生物泛基因组分析的高效工具,专门设计用于处理来自不同菌株的基因组注释数据。该工具通过图论方法整合和校正基因注释,能够有效识别和纠正注释错误、检测结构变异,并重构准确的泛基因组。Panaroo支持多种输入格式(GFF3、GenBank等),提供严格、中等和敏感三种运行模式,适应不同质量数据的需求。主要功能包括基因聚类、缺失基因重发现、并行处理、核心基因组对齐等。与传统工具相比,Panaroo在保持高精度的同时,显著提高了对注释错误的鲁棒性,特别适用于大规模微生物基因组比较研究。
文章介绍
研究背景与问题陈述
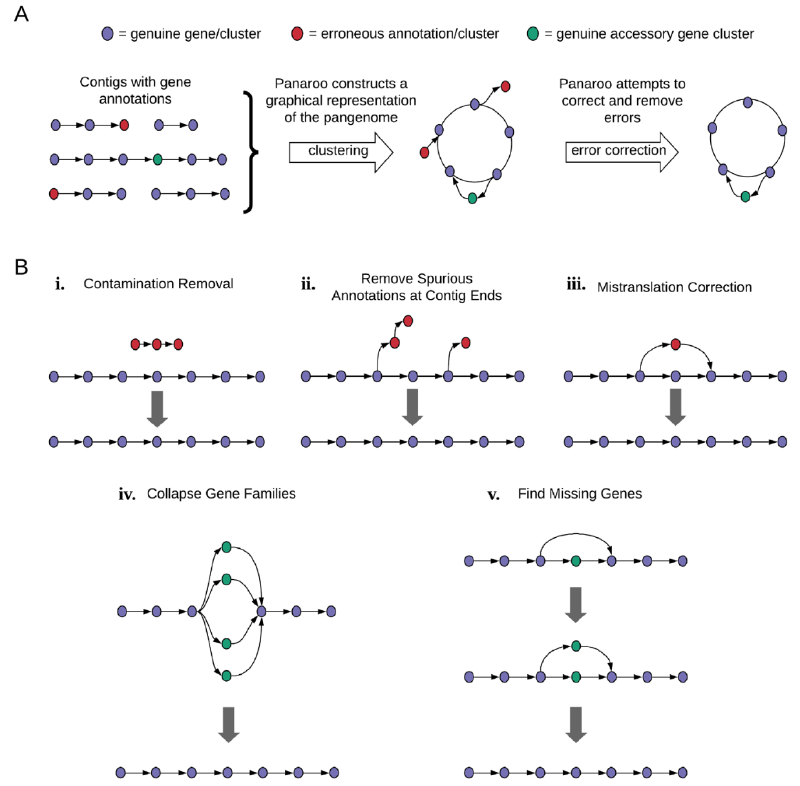
微生物基因组学研究经常涉及多个菌株的比较分析,传统的泛基因组分析工具(如Roary)在处理大规模数据集时面临诸多挑战。注释软件(如Prokka)在不同基因组中可能对相同基因序列产生不一致的注释,导致基因聚类错误。此外,基因组组装碎片化、测序错误和污染等问题进一步增加了分析的复杂性。Panaroo的开发旨在解决这些挑战,通过创新的图论算法提供更准确的泛基因组表征。
研究方法与技术路线

Panaroo采用多阶段处理流程,核心算法包括:
- 初始基因聚类:使用CD-HIT进行保守聚类,默认参数为98%序列相似性和98%长度差异阈值
- 图构建:将基因聚类表示为图结构,节点代表基因家族,边代表基因组中的相邻关系
- 图校正:通过多种启发式方法识别和校正注释错误,包括:
- 末端基因修剪:移除低支持度的末端簇
- 边缘过滤:基于支持度阈值清理可疑连接
- 长度异常值检测:处理长度异常的基因注释
- 基因重发现:在注释缺失的基因组区域搜索可能存在的基因
- 并行化处理:支持多线程加速,提高大规模数据集处理效率
主要发现与创新点
Panaroo的主要创新体现在以下几个方面:
- 智能错误校正:通过图论方法系统识别和校正多种类型的注释错误,显著提高聚类准确性
- 灵活的运行模式:提供strict、moderate、sensitive三种模式,适应不同数据质量需求
- 基因重发现功能:能够检测和恢复被注释工具遗漏的基因,提高基因检测灵敏度
- 结构变异分析:内置结构变异检测功能,可直接从泛基因组图中识别基因组重排事件
- 可视化支持:生成兼容Cytoscape的图文件,便于结果可视化和解释
研究意义与贡献
Panaroo为微生物比较基因组学研究提供了更可靠的分析工具,特别是在处理大规模、多样化数据集时表现出色。其准确的基因聚类和错误校正能力有助于发现真实的基因含量差异,为病原菌进化研究、功能基因组学和疫苗设计等领域提供更可靠的数据基础。工具的开源特性也促进了方法的透明性和可重复性。
使用教程
安装部署
环境要求
- Python 3.9或更高版本(Bioconda目前推荐使用3.9)
- 至少4GB内存(大型数据集需要更多)
- 足够的磁盘空间存储中间文件和结果
依赖安装
Conda安装(推荐)
|
|
手动安装
|
|
可选依赖
- Prokka:用于基因组注释
- PRANK/Clustal:替代MAFFT进行多序列比对
- MASH:用于质量控制和参考数据库比较
基本使用方法
快速开始
|
|
处理RefSeq格式文件
RefSeq注释文件可能包含不符合预期的注释,如包含提前终止密码子的基因:
|
|
运行模式选择
|
|
输入格式支持
Panaroo支持多种输入格式:
- 标准GFF3文件
- 包含文件路径列表的文本文件
- 分隔的GFF和FASTA文件
- GenBank格式文件(.gbk, .gb, .gbff)
进阶应用
多序列比对生成
|
|
基因家族聚类参数调整
|
|
处理旁系同源基因
|
|
基因重发现参数优化
|
|
数据预处理和质量控制
|
|
参考 mash 数据库可以从 https://mash.readthedocs.io/en/latest/tutorials.html 下载
一致性注释生成
|
|
参数详解
核心参数说明
--clean-mode:运行模式,影响错误校正的严格程度--threshold:初始聚类序列相似性阈值(默认0.98)--family_threshold:基因家族聚类阈值(默认0.7)--core_threshold:核心基因组定义阈值(默认0.95)
不同模式的默认参数
| 参数 | 严格模式 | 中等模式 | 敏感模式 |
|---|---|---|---|
| min_trailing_support | max(2, ⌈0.05N⌉) | max(2, ⌈0.01N⌉) | 2 |
| trailing_recursive | ∞ | ∞ | 1 |
| remove_by_consensus | True | False | False |
常见问题与解决方案
安装问题
问题1:Python版本兼容性错误
|
|
问题2:依赖包冲突
|
|
运行错误
问题1:无效基因注释错误
|
|
问题2:内存不足
|
|
性能优化
- 大型数据集(>100基因组)建议使用8-16线程
- 敏感模式需要更多内存和计算时间
- 预处理阶段使用panaroo-qc识别潜在问题
输出文件详解
主要输出文件
gene_presence_absence.csv
基因存在/不存在矩阵,格式与Roary兼容。包含每个基因簇在各样本中的存在情况,合并的注释用分号分隔,含终止密码子的基因添加"_stop"后缀,长度异常基因添加"_len"后缀。
gene_presence_absence.Rtab
二进制版本的基因存在/不存在矩阵,便于统计分析。
final_graph.gml
最终泛基因组图,可使用Cytoscape可视化或NetworkX进行进一步分析。包含基因注释和基因组特异性信息。
struct_presence_absence.csv
基因组重排事件的存在/不存在矩阵,可通过--min_edge_support_sv参数调整检测阈值。
pan_genome_reference.fa
线性参考基因组,包含数据集中所有基因。注意:旁系同源基因簇仅出现一次以避免多映射问题。
序列文件
combined_DNA_CDS.fasta
包含所有注释和重发现基因的核苷酸序列,使用Panaroo内部命名,可通过gene_data.csv文件转换为原始名称。
combined_protein_CDS.fasta
与上述文件对应的蛋白质序列。
比对文件
core_gene_alignment.aln
核心基因多序列比对文件,包含至少达到--core_threshold阈值菌株中存在的基因。
core_gene_alignment_filtered.aln
过滤后的核心基因组比对,推荐用于构建系统发育树。使用BMGE过滤异常基因,默认基于Tukey异常值检测自动调整。
References
- Tonkin-Hill, G., MacAlasdair, N., Ruis, C., Weimann, A., Horesh, G., Lees, J.A., Gladstone, R.A., Lo, S., Beaudoin, C., Floto, R.A., et al. (2020). Producing polished prokaryotic pangenomes with the panaroo pipeline. Genome Biol. 21, 180. https://doi.org/10.1186/s13059-020-02090-4.
- https://gthlab.au/panaroo/