Introduction
最近有审稿人的comments中建议阅读大佬Susan Holmes (2014) 的经典论文《Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible》,参考在自己的分析中。这篇文章深入探讨了微生物组数据分析中常用的稀释(rarefying)方法的缺陷,并提出了更合适的统计模型。虽然这篇文章已经发表了10多年,但其观点仍然具有重要的参考价值。
🌱 微生物组测序的统计挑战
高通量 DNA 测序技术让微生物组研究进入爆发期。通过测定来自不同环境或样本(如人体肠道、土壤、水体)的 16S rRNA 序列或宏基因组原始读数,研究者可以获得一个 OTU(操作分类单位,又可视作微生物“物种”)在各样本中的丰度矩阵。
但一个关键问题是:不同样本之间测序深度存在显著差异——每个样本的读数(library size)可能相差一个数量级。这样直接比较 OTU 计数会导致误导。为此,研究者常用两种“归一化”方式:
- 按比例(proportion)处理:将各 OTU 计数除以总读数。
- 稀释抽平(rarefying):随机抽取每个样本相同数量的读数(即归到最小 library size)。
然而,McMurdie 和 Holmes 指出,这两种方法都存在严重统计缺陷。作者主张,应该使用更先进的统计模型,比如基于负二项分布的差异表达分析方法(edgeR、DESeq),这些方法能更好地处理 library size 差异和数据方差结构。
稀释法的两个主要问题
损失统计能力(statistical power)

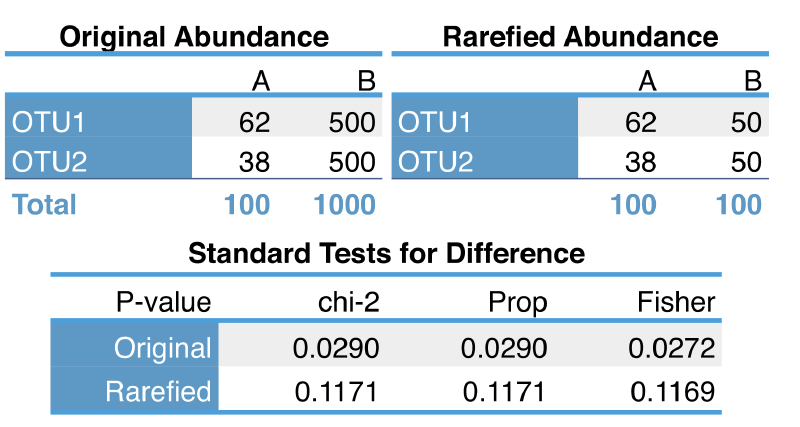
论文通过例子演示了 rarefying 带来的信息损失,例如样本 B 从 1000 个读数降到 100 后,估计微生物丰度比例的方差显著增加,导致对显著性差异的检验能力下降。即使两个样本在真实生态差异上有明显变化,稀释后信号可能被掩盖。
丢弃样本
在 rarefying 流程中,通常设定一个最小读数阈值,低于阈值的样本被剔除。例如,如果用 500 读数标准化,而一个 sample 只有 300,则该 sample 会被完全删除。论文中指出,这可能导致有效样本被剔除,从而降低聚类分析或差异检测的结果质量。
替代方案:基于负二项分布的混合模型
论文提出将微生物组计数数据视作负二项分布(Negative Binomial, NB)的一种表现:
- 样本 j 的总体读数可视为 Poisson 随机变量。
- OTU i 在样本 j 的计数$K[i,j] ~ NB(mean = s_j × μ_i, dispersion = φ_i)$,其中$s_j$ 是 library size 缩放因子,$μ_i$ 是 OTU 的相对丰度,$φ_i$ 捕捉 biological variability 和过度离散性。
这种模型有三个优点:
- 统计理论支持:standard 方法来自 RNA-Seq 领域(edgeR、DESeq)。
- 变异估计更准确:考虑方差随着丰度增长而变化,避免样本深度不同导致的 heteroscedasticity。
- 没丢样本:所有样本保留,避免信息丢失。
此外还提及另一种方法,zero-inflated Gaussian 模型(metagenomeSeq),适合候选 OTU 零计数很多的情况,但在生物重复较少时假阳性率仍然较高。
模拟实验
McMurdie 和 Holmes 为了系统地评估不同归一化和统计分析方法的性能,精心设计了两类模拟实验,分别聚焦于 样本聚类准确性 与 差异丰度检出能力。这些实验通过真实数据构造,并在可控的干预条件下测试各方法的稳健性与准确度,具有非常强的说服力。

A:样本聚类的准确性(Clustering Accuracy)
作者从真实的微生物组数据集中(如“soil”和“ocean”数据)选取样本,构建两个组成明显不同的样本群体,并在这些群体中模拟不同的测序深度(library size),例如一个样本有 10,000 reads,而另一个只有 1,000 reads,反映现实中常见的不平衡情况。
接着,他们使用不同的归一化方式处理这些样本,包括:
- 稀释(rarefying):所有样本统一稀释至相同深度。
- 比例归一化(proportions):每个 OTU 相对其样本总数转换为相对丰度。
- 基于模型的方法(edgeR / DESeq):使用负二项分布估计归一化因子并建模。
- metagenomeSeq:使用零膨胀正态模型(zero-inflated Gaussian)。
归一化后,再对 OTU 表进行距离计算(Bray-Curtis 或 UniFrac),最后进行聚类(如 PAM 或 hierarchical clustering)以观察样本是否能准确归类。

- 稀释法准确率最低,尤其当稀释程度较高(即大部分样本初始 read 数远高于稀释阈值)时,稀释过程导致数据大幅减少,聚类准确率明显下降;
- 比例归一化方法略好但仍不稳定,尤其在物种丰富度差异大时,常低估丰度低但重要的 OTU;
- edgeR 和 DESeq 准确率最高且最稳定,能有效识别样本之间真实的结构差异;
- metagenomeSeq 表现介于中间,在某些条件下表现尚可,但 replicates 少时不稳定。
这说明:基于模型的方法不仅能保留样本完整性,还能在复杂多变的测序深度下维持稳定的聚类表现。
B:差异丰度检测能力(Differential Abundance Detection)
作者构建了一个 OTU 表,其中一部分 OTU 被设定为“差异丰度 OTU”——它们在组 A 与组 B 中的丰度存在系统性差异(如增加 2 倍或 5 倍),而其余 OTU 丰度一致不变。
通过不同方法处理这些数据并检测差异 OTU,然后使用 ROC 曲线和 AUC(Area Under Curve) 来衡量每种方法检出真实差异 OTU 的能力与假阳性控制水平。

edgeR 与 DESeq 明显优于其他方法,具体表现为:
- AUC 高达 0.95–0.98,说明检出真正差异 OTU 的能力极强;
- 假阳性率控制良好,尤其在样本数较多时尤为明显;
- 稀释法表现最差,不仅灵敏度低(漏检多),而且假阳性率高,尤其当稀释阈值较低时更严重;
- metagenomeSeq 在 replicates 较多时接近 edgeR,但在 n=2~3 样本时性能急剧下降。
此外,作者强调,稀释法带来的最大问题是“有效样本数的减少”——本来可以使用 10,000 个 reads 做差异分析,稀释后只剩 1,000 个,等效于用更小的样本重复去估计参数,导致功效大幅下降。
McMurdie & Holmes 在文中总结如下:
- 明确反对 rarefying:既浪费数据又带来分析偏差,是 inadmissible 的 normalization 方法。
- 推荐基于统计混合模型的方法:借用 RNA-Seq 的 edgeR、DESeq,甚至 metagenomeSeq。作者还将这些方法整合进 R 包 phyloseq 中,方便微生物组分析。
- 树立统计意识:研究者应理解 library size、overdispersion 的本质,并采用合适的 GLM(Generalized Linear Model)模型或经验贝叶斯方法,而不是简单采样。
这篇论文自 2014 年发布后对微生物组数据分析流程影响深远:
- 推动了差异丰度分析方法的演进,例如 ANCOM、ALDEx2、Songbird 等工具开始强调 compositional data 和统计模型。
- 2024 年,Schloss 又在 mSphere 中重访 rarefying,提出一些更新观点,可视为对这篇论文的回应和延伸。
- 微生物组数据本质是成分数据(compositional),总和约束 → 需要特殊方法处理,而 NB 模型正是合理起点之一。
References
- McMurdie, P.J., and Holmes, S. (2014). Waste not, want not: why rarefying microbiome data is inadmissible. PLOS Comput. Biol. 10, e1003531. https://doi.org/10.1371/journal.pcbi.1003531.