在复杂网络分析中,模块(module)或社区(community)是指网络中连接更为紧密的子图结构。这些模块通常代表功能相关的节点群组,在生物网络中可能对应特定的功能单元或调控模块。MetaNet工具包提供了全面的功能,本文将详细介绍其核心方法和应用场景。
可以从 CRAN 安装稳定版:install.packages("MetaNet")
依赖包 pcutils和igraph(需提前安装),推荐配合 dplyr 进行数据操作。
1
2
3
4
5
6
|
library(MetaNet)
library(igraph)
# ========data manipulation
library(dplyr)
library(pcutils)
|
网络模块(module)
模块(module)或社区(community)是指包含节点的子图,其中节点之间的连接密度高于它们与图中其他节点的连接密度。用数学语言表达:当任何子图内部的连接数高于这些子图之间的连接数时,我们就说这个图具有社区结构。
在MetaNet中,可以使用module_net()函数生成具有指定模块数的网络:
1
2
3
4
|
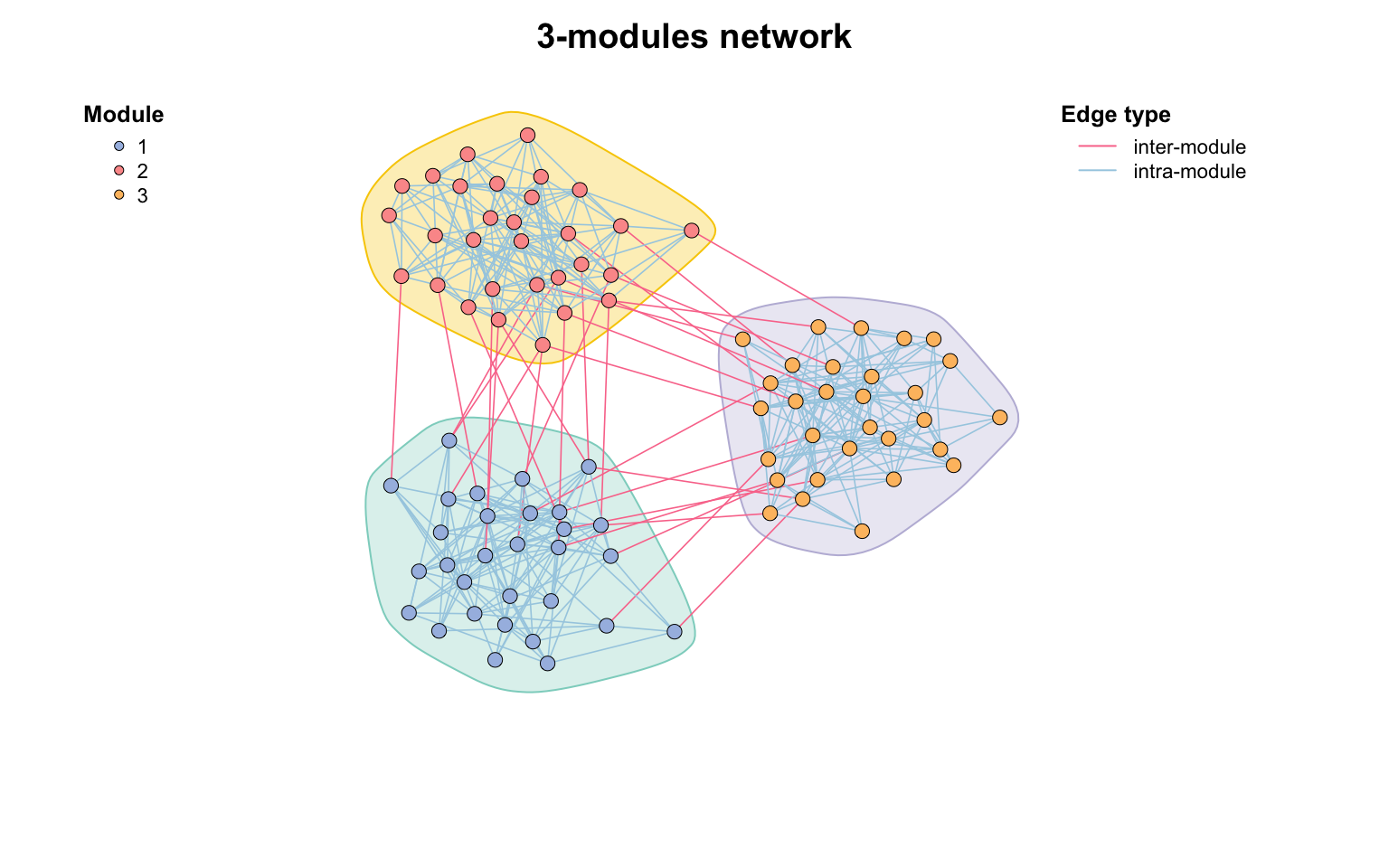
set.seed(12)
# 生成包含3个模块的网络,每个模块30个节点
test_module_net <- module_net(module_number = 3, n_node_in_module = 30)
plot(test_module_net, mark_module = TRUE)
|
网络科学领域已开发出多种模块检测算法,各有其优势和适用场景:
- 短随机游走法:基于随机游走的动态过程识别社区
- 社区矩阵的主特征向量法:利用矩阵特征向量进行谱聚类
- 模拟退火法:通过优化模块度指标寻找全局最优解
- 贪婪模块度优化:局部搜索算法,计算效率较高
…
MetaNet的module_detect()函数集成了这些主流算法,用户可以根据网络特性选择合适的方法。对于大型网络,建议先测试不同算法的运行时间和效果。
1
2
|
# 使用快速贪婪算法检测模块
module_detect(co_net, method = "cluster_fast_greedy") -> co_net_modu
|
模块筛选合并
实际分析中,我们常关注特定规模的模块。filter_n_module()函数支持多种筛选方式:
• 按节点数筛选:保留节点数超过阈值的模块
• 按模块ID筛选:指定需要保留的特定模块
• 组合筛选:同时应用多种条件
网络布局对模块展示效果至关重要,之前介绍了很多g_layout方法都可以在这里用上了。g_layout_circlepack()可生成基于模块的圆形堆积布局:
1
2
3
4
|
par(mfrow = c(2, 2), mai = rep(1, 4))
# module detection
module_detect(co_net, method = "cluster_fast_greedy") -> co_net_modu
get_v(co_net_modu)[, c("name", "module")] %>% head()
|
1
2
3
4
5
6
7
|
## name module
## 1 s__un_f__Thermomonosporaceae 10
## 2 s__Pelomonas_puraquae 9
## 3 s__Rhizobacter_bergeniae 1
## 4 s__Flavobacterium_terrae 3
## 5 s__un_g__Rhizobacter 14
## 6 s__un_o__Burkholderiales 9
|
1
2
3
4
5
|
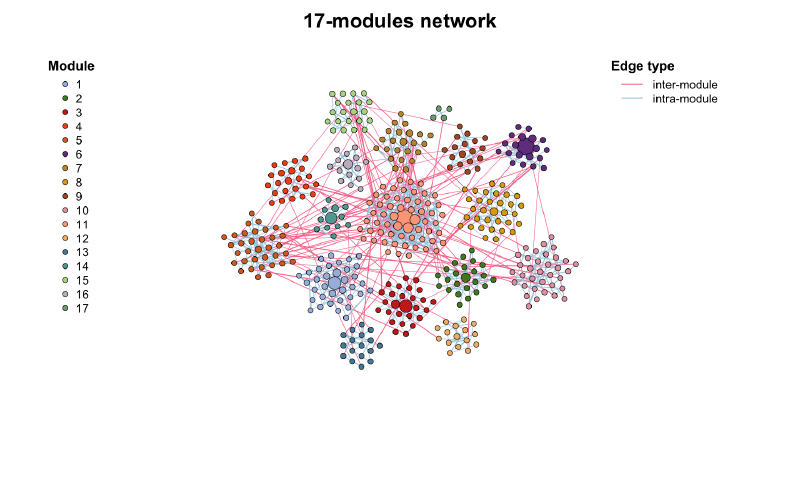
plot(co_net_modu,
plot_module = T, mark_module = T,
legend_position = c(-1.8, 1.6, 1.1, 1.3), edge_legend = F
)
table(V(co_net_modu)$module)
|
1
2
3
4
5
|
##
## 1 10 11 12 13 14 15 16 17 18 19 2 20 21 22 23 24 25 26 27 28 29 3 4 5 6
## 36 18 35 16 17 12 21 15 6 4 4 24 2 3 2 2 2 2 3 2 3 2 27 23 35 23
## 7 8 9
## 33 18 61
|
1
2
3
4
5
6
7
8
9
10
11
|
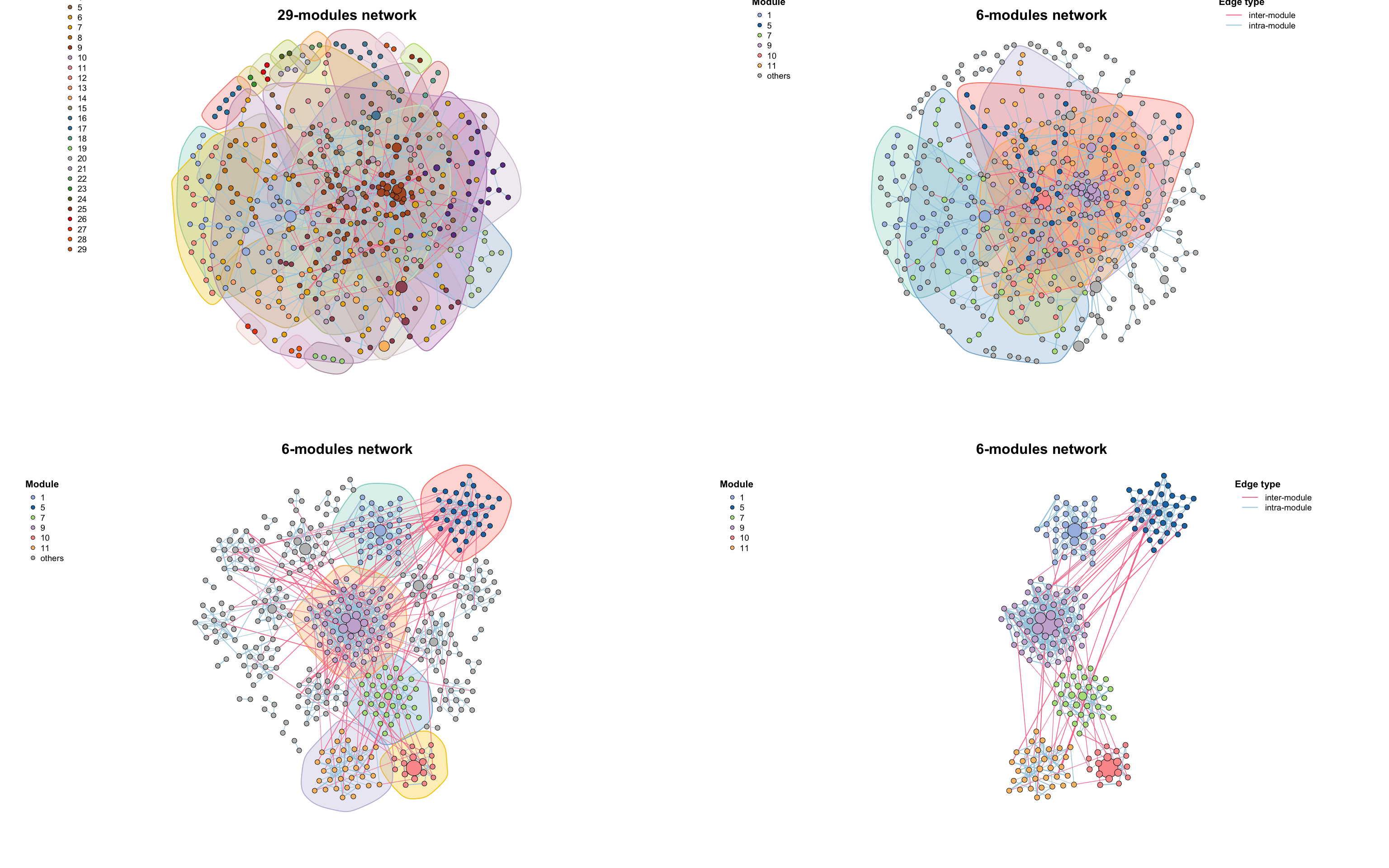
# 保留节点数≥30的模块和ID为10的模块
co_net_modu2 <- filter_n_module(co_net_modu, n_node_in_module = 30, keep_id = 10)
plot(co_net_modu2, plot_module = T, mark_module = T, legend_position = c(-1.8, 1.3, 1.1, 1.3))
# change group layout
g_layout_circlepack(co_net_modu, group = "module") -> coors
plot(co_net_modu2, coors = coors, plot_module = T, mark_module = T, edge_legend = F)
# extract some modules, delete =T will delete other modules.
co_net_modu3 <- filter_n_module(co_net_modu, n_node_in_module = 30, keep_id = 10, delete = T)
plot(co_net_modu3, coors, plot_module = T)
|
看看网络的components,一些太小的sub_graphs会影响模块,如果您不关心这些小型组件,则可以过滤掉它们。
1
|
table(V(co_net_modu)$components)
|
1
2
3
|
##
## 1 10 11 12 13 2 3 4 5 6 7 8 9
## 418 2 2 2 2 6 4 2 2 3 2 3 3
|
1
2
3
4
5
6
|
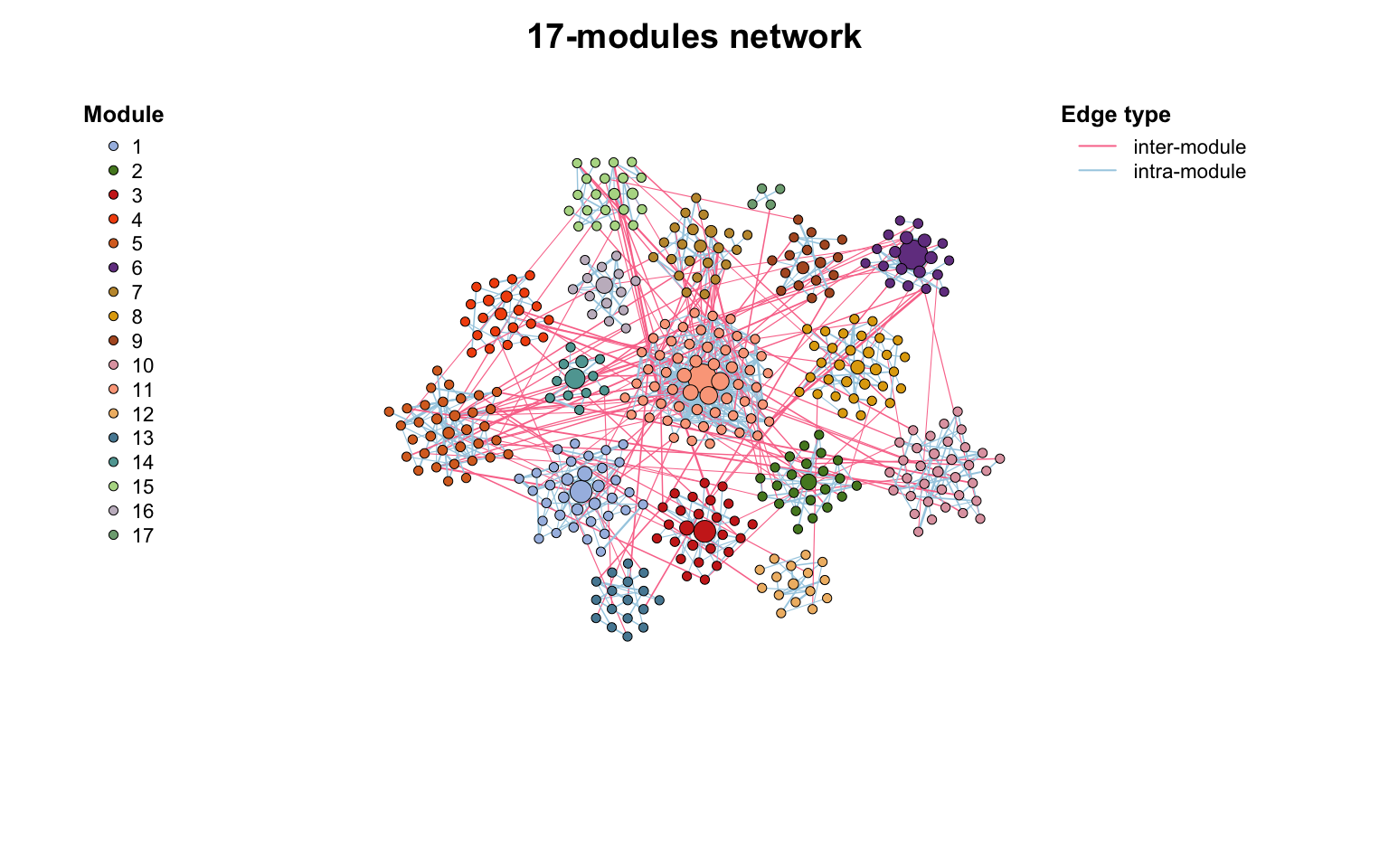
co_net_modu4 <- c_net_filter(co_net_modu, components == 1)
# re-do a module detection
co_net_modu4 <- module_detect(co_net_modu4)
g_layout_circlepack(co_net_modu4, group = "module") -> coors
plot(co_net_modu4, coors, plot_module = T)
|
plot_module_tree()函数可展示模块的树状关系,揭示模块间的层次结构。当模块数量过多时,combine_n_module()可将模块合并到指定数量,便于高层次分析。
1
2
3
4
5
6
7
8
9
|
# 展示模块树状图
p1 <- plot_module_tree(co_net_modu4, label.size = 0.6)
# 将17个模块合并为5个
co_net_modu5 <- combine_n_module(co_net_modu4, 5)
p2 <- plot_module_tree(co_net_modu5, label.size = 0.6)
library(patchwork)
p1+p2
|
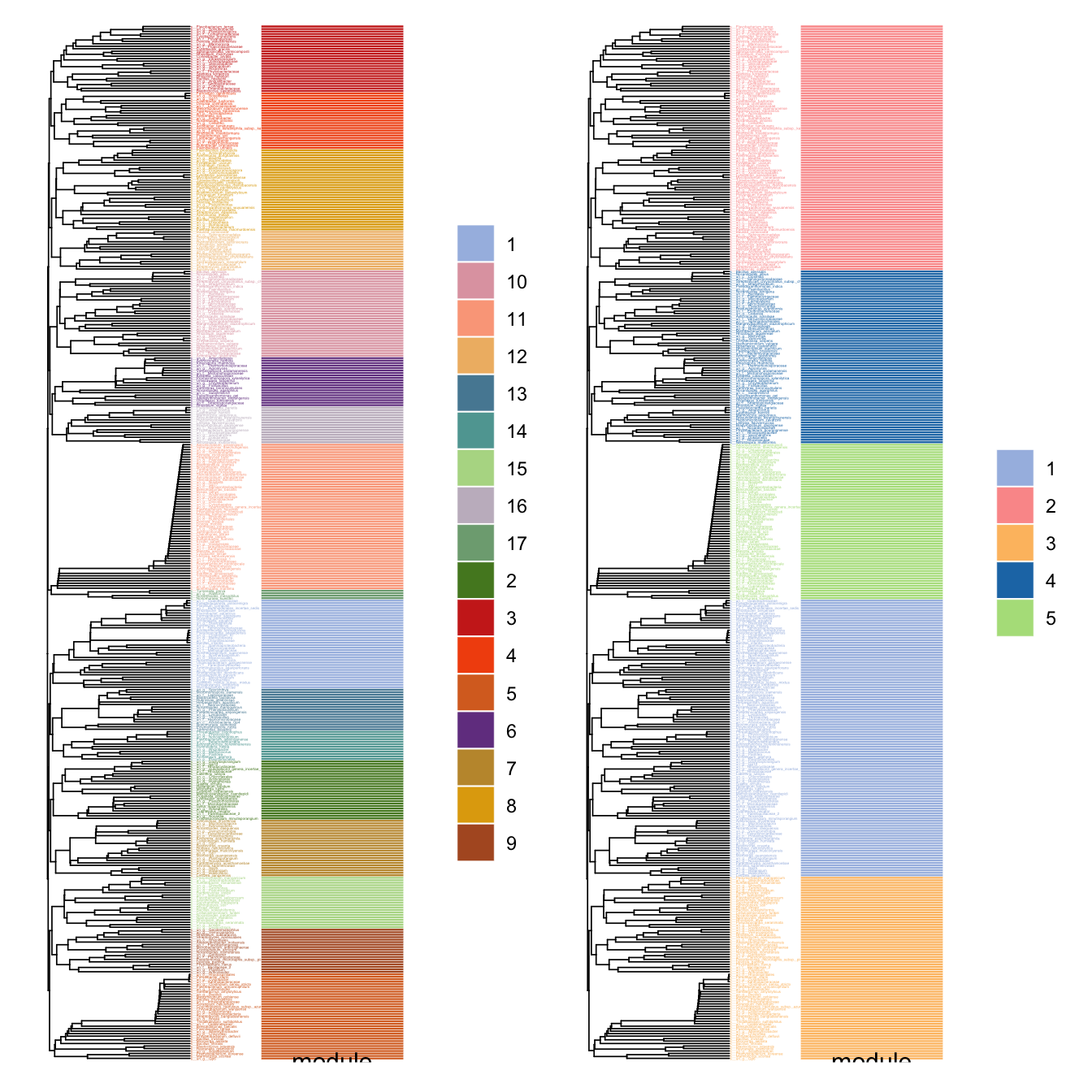
模块pattern分析
在生物网络中,模块常对应功能相关的分子集合。我们还可以使用此网络模块指示具有相似表达/丰度的群集。但是我们应该首先过滤正边,因为模块检测仅考虑拓扑结构而不是边缘类型。过滤正相关边和模块检测后,将找到一些模块,很像是WGCNA里的基因模块,我们还可以使用module_eigen查看每个模块表达模式。
1
2
3
4
5
6
7
8
|
data("otutab", package = "pcutils")
totu <- t(otutab)
# filter positive edges
c_net_filter(co_net, e_type == "positive", mode = "e") -> co_net_pos
co_net_pos_modu <- module_detect(co_net_pos, n_node_in_module = 15, delete = T)
g_layout_circlepack(co_net_pos_modu, group = "module") -> coors1
plot(co_net_pos_modu, coors1, plot_module = T)
|
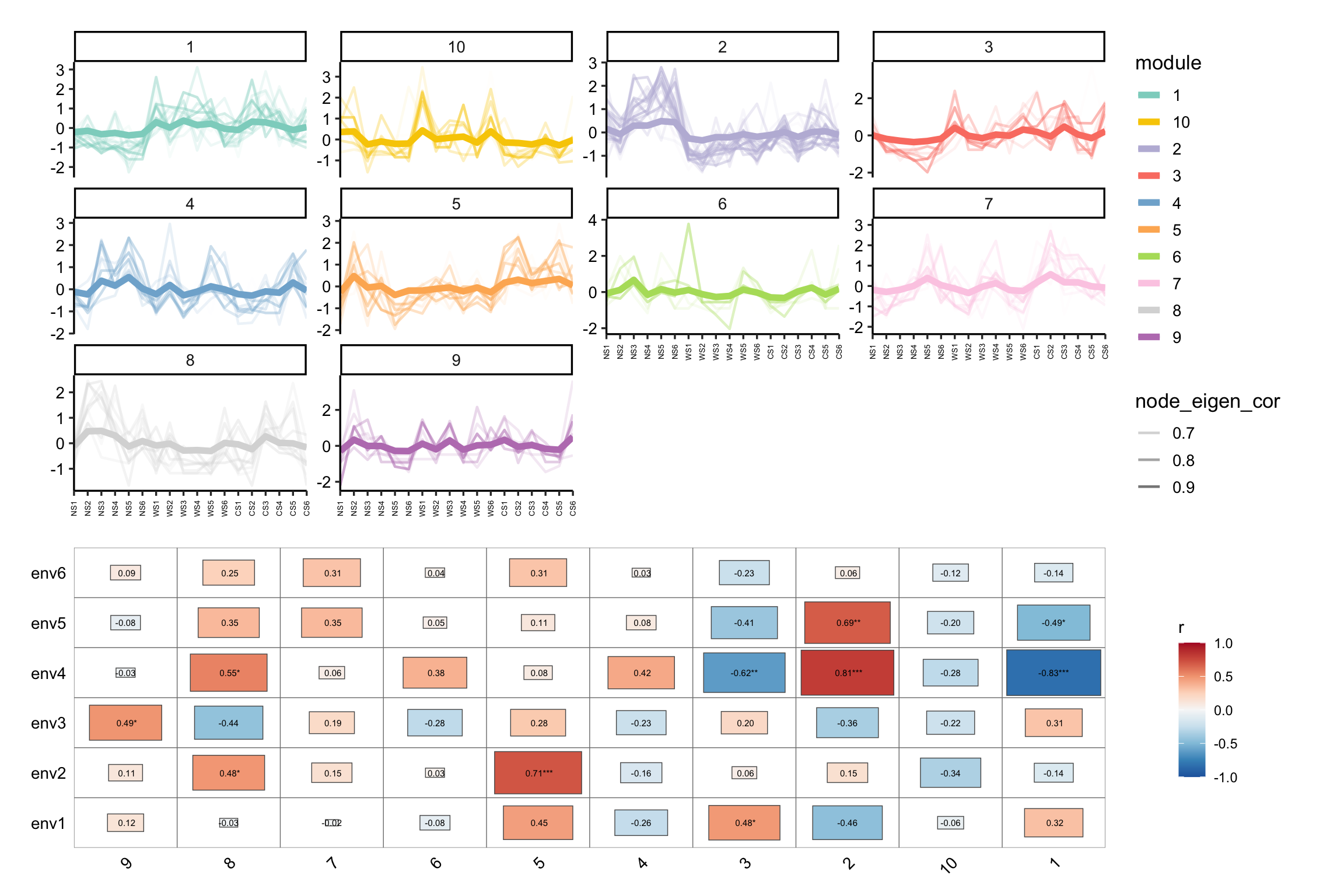
module_eigen()和module_expression()可计算和可视化模块特征基因及表达模式:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
# map the original abundance table
module_eigen(co_net_pos_modu, totu) -> co_net_pos_modu
# plot the expression pattern
p1 <- module_expression(co_net_pos_modu, totu,
r_threshold = 0.6,
facet_param = list(ncol = 4), plot_eigen = T
) +
theme(axis.text.x = element_text(size = 5, angle = 90, vjust = 0.5))
# correlate to metadata
env <- metadata[, 3:8]
p2 <- cor_plot(get_module_eigen(co_net_pos_modu), env) + coord_flip()
p1 / p2 + patchwork::plot_layout(heights = c(2, 1.4))
|
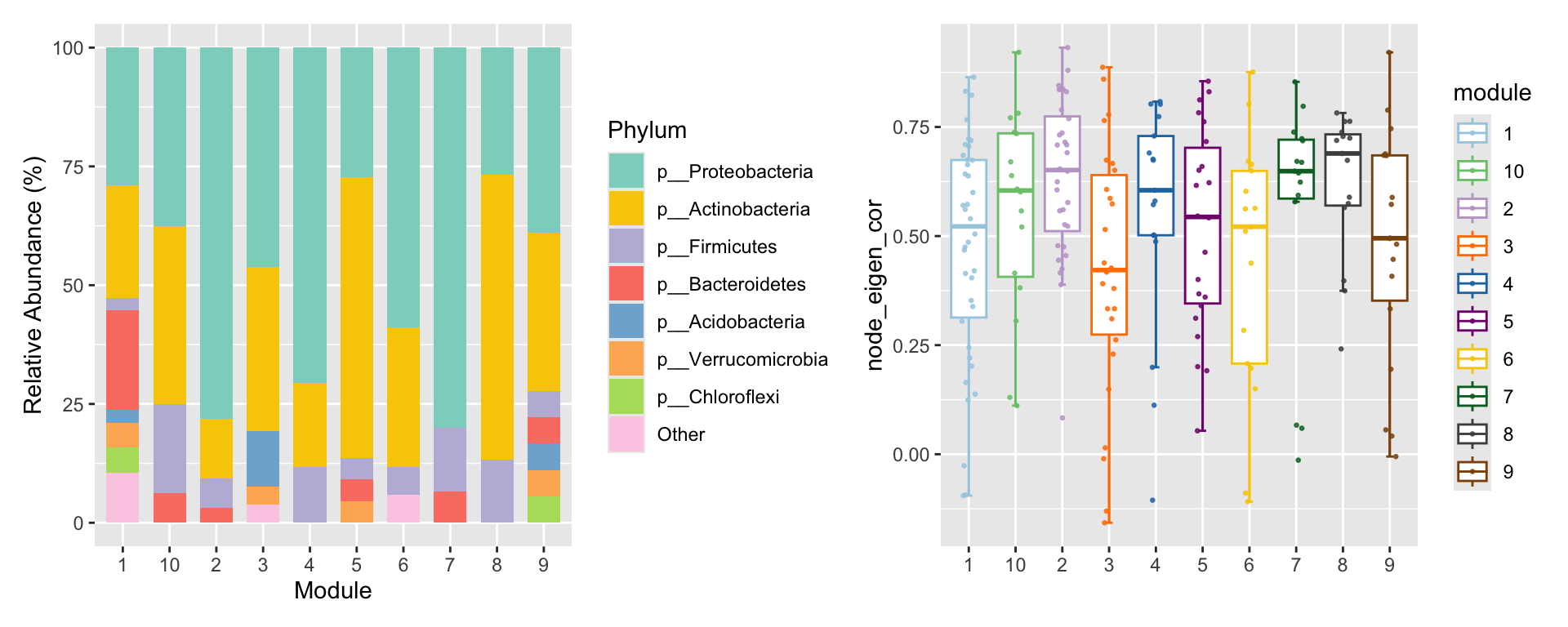
1
2
3
4
5
6
|
# summary some variable according to modules.
p3 <- summary_module(co_net_pos_modu, var = "Phylum") +
scale_fill_pc()
p4 <- summary_module(co_net_pos_modu, var = "node_eigen_cor") +
scale_color_pc(palette = "col2")
p3 + p4
|
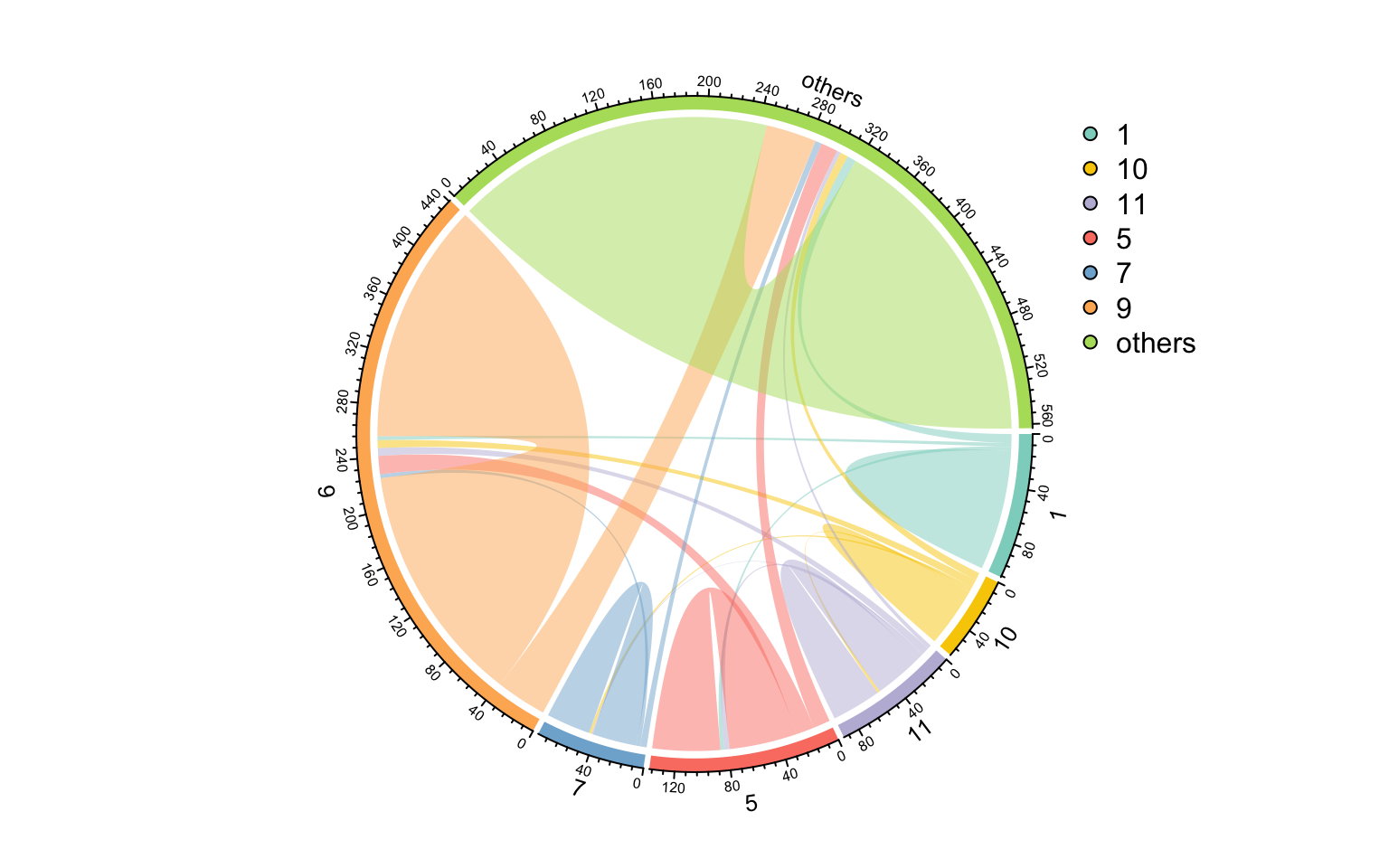
使用links_stat()对边进行汇总,发现大多数边都是从一个模块到同一个模块的(意味着模块检测正常)。
1
|
links_stat(co_net_modu2, group = "module")
|
拓扑角色分析
在我们确定了网络的这些模块后,可以根据Zi-Pi计算每个节点的拓扑角色。
Within-module connectivity (Zi):
$$
Z_i= \frac{\kappa_i-\overline{\kappa_{si}}}{\sigma_{\kappa_{si}}}
$$
其中$κ_i$是节点i到其模块si中其他节点的链接数,$\overline{\kappa_{si}}$是si中所有节点的k的平均值,$\sigma_{\kappa_{si}}$是si中κ的标准偏差。
Among-module connectivity (Pi):
$$
P_i=1-\sum_{s=1}^{N_m}{\left( {\frac{\kappa_{is}}{k_i}} \right)^2}
$$
其中$\kappa_{is}$是节点i到模块s中节点的链接数,$k_i$是节点i的总度数。
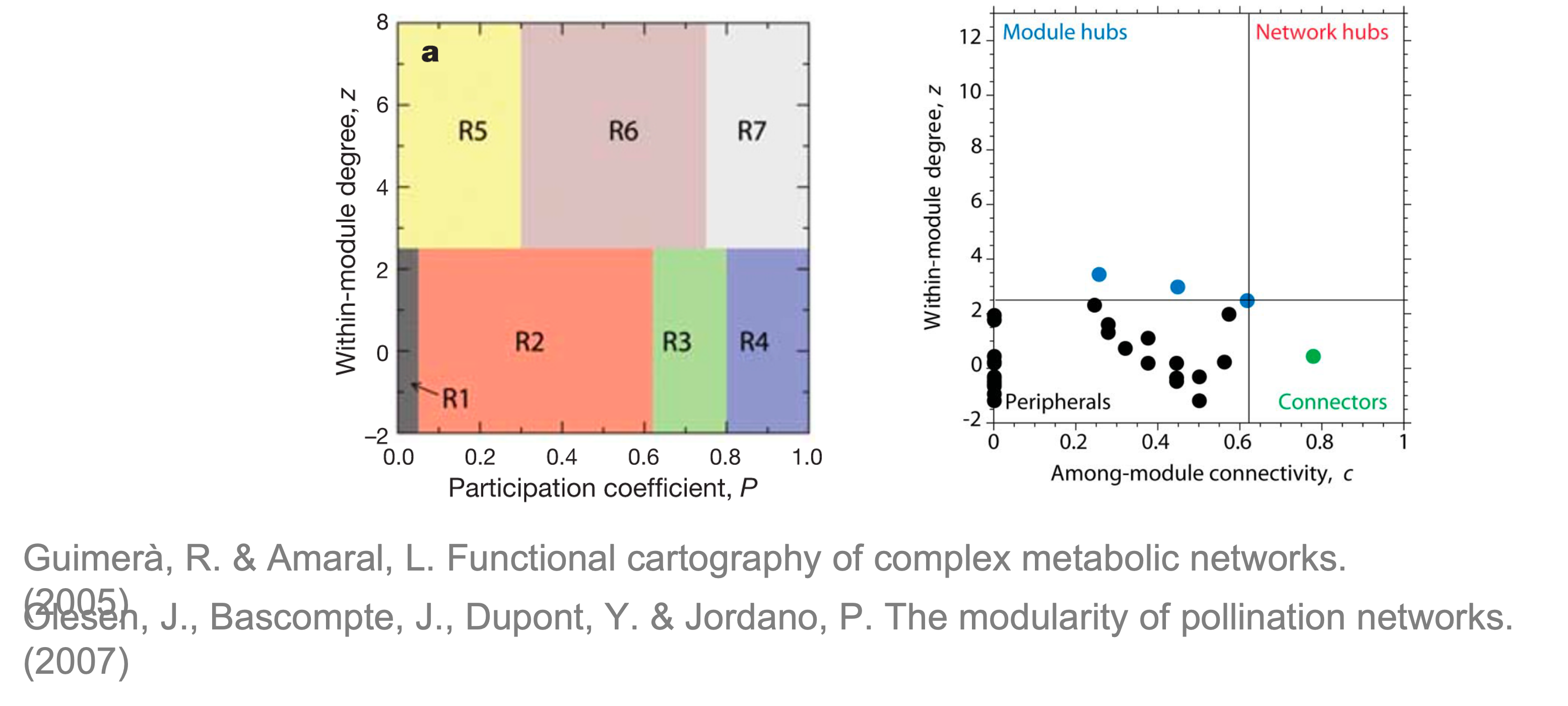
参考R. Guimerà, L. Amaral, Functional cartography of complex metabolic networks (2005), doi:10.1038/nature03288.,基于Zi-Pi指标,节点可分为四类拓扑角色:
- 外围节点(Peripherals):Zi<2.5且Pi<0.62
- 模块枢纽(Module hubs):Zi>2.5且Pi<0.62
- 连接节点(Connectors):Zi<2.5且Pi>0.62
- 网络枢纽(Network hubs):Zi>2.5且Pi>0.62
其中除了Peripherals的节点通常被视为网络的关键节点(keystone),参考
S. Liu, H. Yu, Y. Yu, J. Huang, et al., Ecological stability of microbial communities in Lake Donghu regulated by keystone taxa. Ecological Indicators. 136, 108695 (2022).
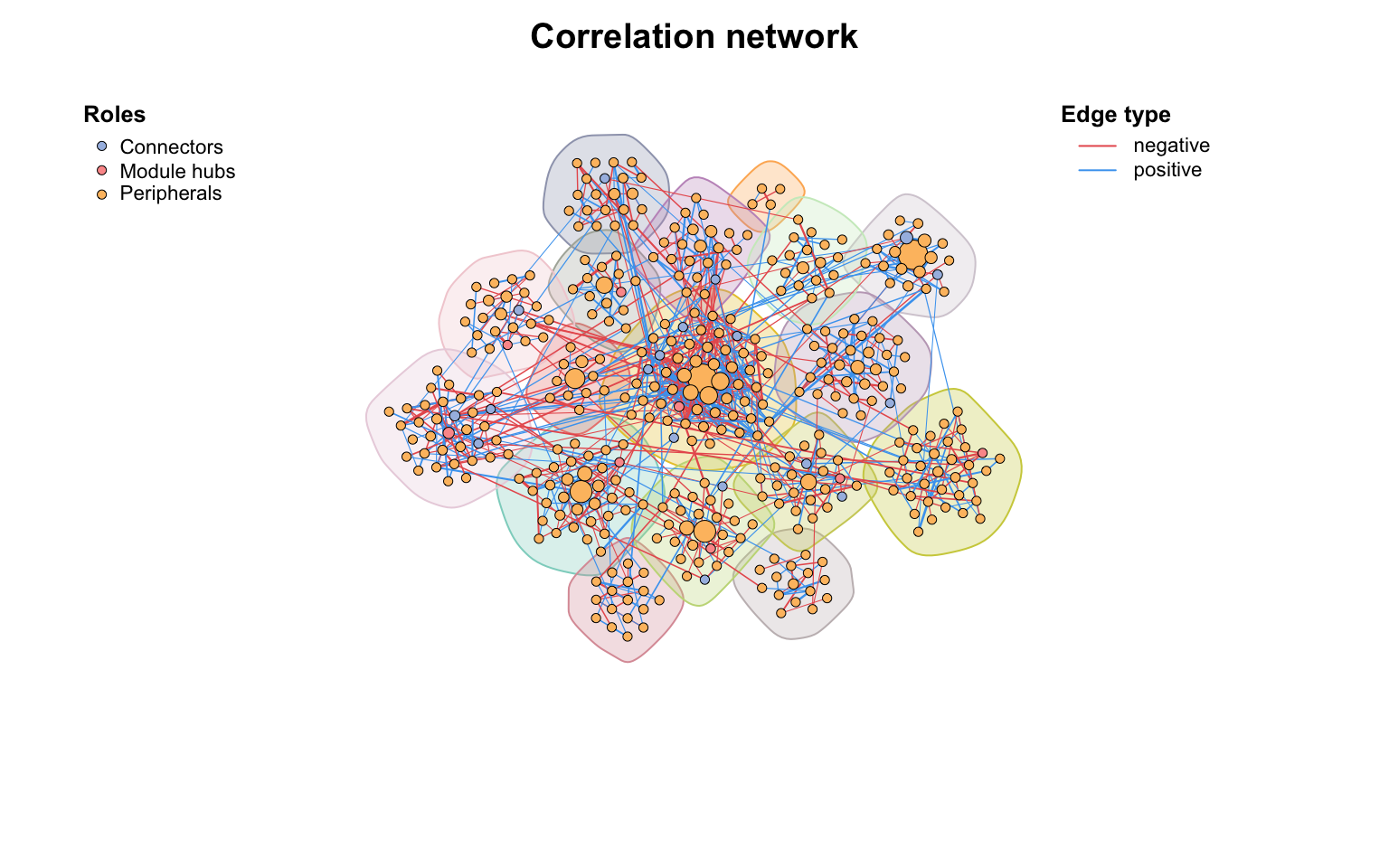
使用zp_analyse拿到模块角色并存储在顶点属性中,然后我们可以使用zp_plot()可视化。我们可以看到模块中心是模块的中心,而连接器通常是介导不同模块的连接。
1
2
|
zp_analyse(co_net_modu4) -> co_net_modu4
get_v(co_net_modu4)[, c(1, 16:21)] %>% head()
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
## name components module original_module Ki Zi
## 1 s__un_f__Thermomonosporaceae 1 6 6 3 0.4358899
## 2 s__Pelomonas_puraquae 1 11 11 15 1.9019177
## 3 s__Rhizobacter_bergeniae 1 1 1 4 1.0951304
## 4 s__Flavobacterium_terrae 1 3 3 4 1.8027756
## 5 s__un_g__Rhizobacter 1 14 14 1 -1.0488088
## 6 s__un_o__Burkholderiales 1 11 11 17 2.3326783
## Pi
## 1 0.3750000
## 2 0.3490305
## 3 0.5714286
## 4 0.0000000
## 5 0.0000000
## 6 0.1939058
|
1
2
3
|
# color map to roles
co_net_modu6 <- c_net_set(co_net_modu4, vertex_class = "roles")
plot(co_net_modu6, coors, mark_module = T, labels_num = 0, group_legend_title = "Roles")
|
1
2
3
|
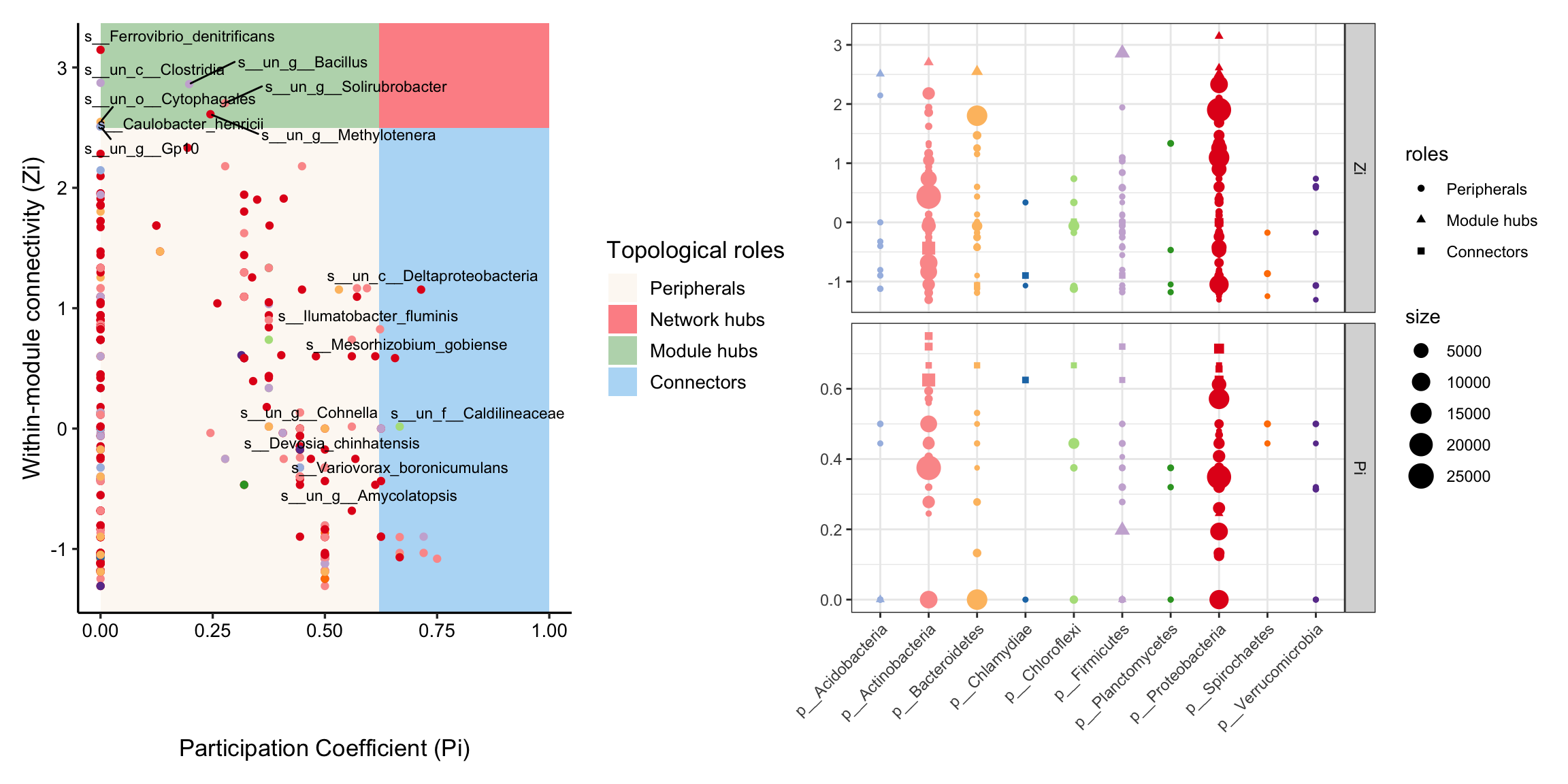
library(patchwork)
zp_plot(co_net_modu4, mode = 1) +
zp_plot(co_net_modu4, mode = 3)
|
References
- Koutrouli M, Karatzas E, Paez-Espino D and Pavlopoulos GA (2020) A Guide to Conquer the Biological Network Era Using Graph Theory. Front. Bioeng. Biotechnol. 8:34. doi: 10.3389/fbioe.2020.00034
- Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. https://doi.org/10.1038/nrmicro2832.
- Y. Deng, Y. Jiang, Y. Yang, Z. He, et al., Molecular ecological network analyses. BMC bioinformatics (2012), doi:10.1186/1471-2105-13-113.
- R. Guimerà, L. Amaral, Functional cartography of complex metabolic networks (2005), doi:10.1038/nature03288.
- S. Liu, H. Yu, Y. Yu, J. Huang, et al., Ecological stability of microbial communities in Lake Donghu regulated by keystone taxa. Ecological Indicators. 136, 108695 (2022).